Saad Tariq, A. A. Mubarak, Ayash O. Alrashdi, Farida Hamioud, Bushra Kanwal
{"title":"通过密度泛函理论分析解读压力对 PrMnO3 电子、机械和结构特性的影响","authors":"Saad Tariq, A. A. Mubarak, Ayash O. Alrashdi, Farida Hamioud, Bushra Kanwal","doi":"10.1007/s11243-023-00540-z","DOIUrl":null,"url":null,"abstract":"<div><p>This is an investigation on the properties of PrMnO<sub>3</sub> using density functional theory under varying pressure conditions ranging from 0 to 50 GPa. The study includes an analysis of the material's structural, electronic, mechanical, and thermal properties, utilizing the computational power of density functional theory. The Goldschmidt tolerance factor, enthalpy, and elastic stability criteria are used to evaluate the material's stability. The results suggest that the material is stable under these criteria. Furthermore, the optimization of the material is discussed based on the computed properties. The results show that the material exhibits good ferromagnetic and spintronic properties, making it a promising candidate for use in optoelectronic and spintronic devices. Overall, the findings highlight the potential of PrMnO<sub>3</sub> to be a valuable material for these applications, as revealed through the use of density functional theory.</p></div>","PeriodicalId":803,"journal":{"name":"Transition Metal Chemistry","volume":"49 1","pages":"1 - 10"},"PeriodicalIF":1.7000,"publicationDate":"2023-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Deciphering the impact of pressure on the electronic, mechanical, and structural properties of PrMnO3 through density functional theory analysis\",\"authors\":\"Saad Tariq, A. A. Mubarak, Ayash O. Alrashdi, Farida Hamioud, Bushra Kanwal\",\"doi\":\"10.1007/s11243-023-00540-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>This is an investigation on the properties of PrMnO<sub>3</sub> using density functional theory under varying pressure conditions ranging from 0 to 50 GPa. The study includes an analysis of the material's structural, electronic, mechanical, and thermal properties, utilizing the computational power of density functional theory. The Goldschmidt tolerance factor, enthalpy, and elastic stability criteria are used to evaluate the material's stability. The results suggest that the material is stable under these criteria. Furthermore, the optimization of the material is discussed based on the computed properties. The results show that the material exhibits good ferromagnetic and spintronic properties, making it a promising candidate for use in optoelectronic and spintronic devices. Overall, the findings highlight the potential of PrMnO<sub>3</sub> to be a valuable material for these applications, as revealed through the use of density functional theory.</p></div>\",\"PeriodicalId\":803,\"journal\":{\"name\":\"Transition Metal Chemistry\",\"volume\":\"49 1\",\"pages\":\"1 - 10\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2023-09-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Transition Metal Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s11243-023-00540-z\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Transition Metal Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11243-023-00540-z","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
Deciphering the impact of pressure on the electronic, mechanical, and structural properties of PrMnO3 through density functional theory analysis
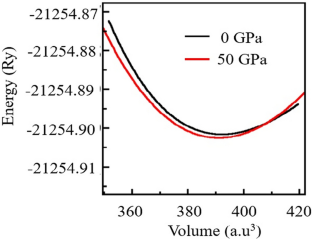
This is an investigation on the properties of PrMnO3 using density functional theory under varying pressure conditions ranging from 0 to 50 GPa. The study includes an analysis of the material's structural, electronic, mechanical, and thermal properties, utilizing the computational power of density functional theory. The Goldschmidt tolerance factor, enthalpy, and elastic stability criteria are used to evaluate the material's stability. The results suggest that the material is stable under these criteria. Furthermore, the optimization of the material is discussed based on the computed properties. The results show that the material exhibits good ferromagnetic and spintronic properties, making it a promising candidate for use in optoelectronic and spintronic devices. Overall, the findings highlight the potential of PrMnO3 to be a valuable material for these applications, as revealed through the use of density functional theory.
期刊介绍:
Transition Metal Chemistry is an international journal designed to deal with all aspects of the subject embodied in the title: the preparation of transition metal-based molecular compounds of all kinds (including complexes of the Group 12 elements), their structural, physical, kinetic, catalytic and biological properties, their use in chemical synthesis as well as their application in the widest context, their role in naturally occurring systems etc.
Manuscripts submitted to the journal should be of broad appeal to the readership and for this reason, papers which are confined to more specialised studies such as the measurement of solution phase equilibria or thermal decomposition studies, or papers which include extensive material on f-block elements, or papers dealing with non-molecular materials, will not normally be considered for publication. Work describing new ligands or coordination geometries must provide sufficient evidence for the confident assignment of structural formulae; this will usually take the form of one or more X-ray crystal structures.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
