Nastaran Pour Ghasem, Robabeh Alizadeh, Sara Seyfi, Vahid Amani
{"title":"镉(II)与 4-甲基-1,2,4-三唑-3-硫醇配体的新型配位聚合物:合成、表征、晶体结构、光致发光和 DFT 计算","authors":"Nastaran Pour Ghasem, Robabeh Alizadeh, Sara Seyfi, Vahid Amani","doi":"10.1007/s11243-023-00564-5","DOIUrl":null,"url":null,"abstract":"<div><p>A new Cadmiun(II) coordination polymer, {[Cd(µ-mptrz)<sub>2</sub>].0.5((CH<sub>3</sub>)<sub>2</sub>SO)}<sub>n</sub> was prepared from the reaction of CdBr<sub>2</sub> with 4-methyl-4<i>H</i>-1,2,4-triazole-3-thiol (Hmptrz) ligand in methanol. The coordination polymer was thoroughly characterized by elemental analysis, IR, UV–Vis, <sup>1</sup>HNMR spectroscopies, and thermogravimetric analysis. Moreover, its structure was studied by the single-crystal diffraction method. Compound <b>1</b> has a 1D chain structure. Furthermore, crystal structure is stabilized by intermolecular weak interactions, for example, C–H···O, C–H···N, and C–H···S hydrogen bonds. Besides, the photoluminescence of <b>1</b> has been investigated. This compound exhibits photoluminescence with an emission maximum of 321 nm upon excitation at 259 nm. The present work is a combined experimental and computational study. Quantum chemical parameters such as bond lengths, bond angles, HOMO–LUMO energy levels, energy band gap <span>\\(\\Delta E\\)</span>, chemical hardness η, the dipole moment μ, and Natural bond orbital (NBO) analysis of the compound were investigated using the DFT at M06/GENECP (6-31 g(d) and LANL2DZ) basis sets for compound <b>1</b>. Our calculation results have shown that compound <b>1</b> has an energy band gap (∆E = 3.857 eV), indicating a high recommendation for a semiconductor compound. The optimized geometry of the Cd(II) coordination polymer is shown in good agreement with single crystal X-ray data. UV/Vis spectra are calculated with the time-dependent density functional theory (TD-DFT) method. Furthermore, TD-DFT displays intraligand transition (ILCT) between triazole bridging ligands. Moreover, NBO analysis of the Cd(II) coordination polymer indicates that the lone pair donor orbital interaction between the N, S, and O lone pairs and π*, σ* anti-bonding orbitals provides stronger stability. Additionally, Solid-state density functional theory (DFT) calculations were performed on <b>1</b> using the program VASP to understand the electronic states and conduction pathways of the coordination polymer.</p></div>","PeriodicalId":803,"journal":{"name":"Transition Metal Chemistry","volume":"49 2","pages":"87 - 100"},"PeriodicalIF":1.7000,"publicationDate":"2023-12-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A new coordination polymer of Cd(II) with 4-methyl-1,2,4-triazole-3-thiol ligand: synthesis, characterization, crystal structure, photoluminescence and DFT calculation\",\"authors\":\"Nastaran Pour Ghasem, Robabeh Alizadeh, Sara Seyfi, Vahid Amani\",\"doi\":\"10.1007/s11243-023-00564-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>A new Cadmiun(II) coordination polymer, {[Cd(µ-mptrz)<sub>2</sub>].0.5((CH<sub>3</sub>)<sub>2</sub>SO)}<sub>n</sub> was prepared from the reaction of CdBr<sub>2</sub> with 4-methyl-4<i>H</i>-1,2,4-triazole-3-thiol (Hmptrz) ligand in methanol. The coordination polymer was thoroughly characterized by elemental analysis, IR, UV–Vis, <sup>1</sup>HNMR spectroscopies, and thermogravimetric analysis. Moreover, its structure was studied by the single-crystal diffraction method. Compound <b>1</b> has a 1D chain structure. Furthermore, crystal structure is stabilized by intermolecular weak interactions, for example, C–H···O, C–H···N, and C–H···S hydrogen bonds. Besides, the photoluminescence of <b>1</b> has been investigated. This compound exhibits photoluminescence with an emission maximum of 321 nm upon excitation at 259 nm. The present work is a combined experimental and computational study. Quantum chemical parameters such as bond lengths, bond angles, HOMO–LUMO energy levels, energy band gap <span>\\\\(\\\\Delta E\\\\)</span>, chemical hardness η, the dipole moment μ, and Natural bond orbital (NBO) analysis of the compound were investigated using the DFT at M06/GENECP (6-31 g(d) and LANL2DZ) basis sets for compound <b>1</b>. Our calculation results have shown that compound <b>1</b> has an energy band gap (∆E = 3.857 eV), indicating a high recommendation for a semiconductor compound. The optimized geometry of the Cd(II) coordination polymer is shown in good agreement with single crystal X-ray data. UV/Vis spectra are calculated with the time-dependent density functional theory (TD-DFT) method. Furthermore, TD-DFT displays intraligand transition (ILCT) between triazole bridging ligands. Moreover, NBO analysis of the Cd(II) coordination polymer indicates that the lone pair donor orbital interaction between the N, S, and O lone pairs and π*, σ* anti-bonding orbitals provides stronger stability. Additionally, Solid-state density functional theory (DFT) calculations were performed on <b>1</b> using the program VASP to understand the electronic states and conduction pathways of the coordination polymer.</p></div>\",\"PeriodicalId\":803,\"journal\":{\"name\":\"Transition Metal Chemistry\",\"volume\":\"49 2\",\"pages\":\"87 - 100\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2023-12-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Transition Metal Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s11243-023-00564-5\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Transition Metal Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11243-023-00564-5","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
A new coordination polymer of Cd(II) with 4-methyl-1,2,4-triazole-3-thiol ligand: synthesis, characterization, crystal structure, photoluminescence and DFT calculation
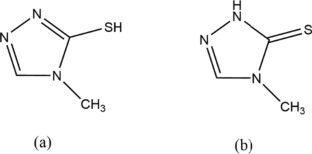
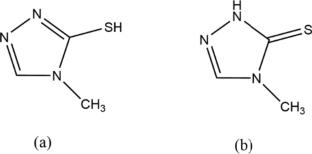
A new Cadmiun(II) coordination polymer, {[Cd(µ-mptrz)2].0.5((CH3)2SO)}n was prepared from the reaction of CdBr2 with 4-methyl-4H-1,2,4-triazole-3-thiol (Hmptrz) ligand in methanol. The coordination polymer was thoroughly characterized by elemental analysis, IR, UV–Vis, 1HNMR spectroscopies, and thermogravimetric analysis. Moreover, its structure was studied by the single-crystal diffraction method. Compound 1 has a 1D chain structure. Furthermore, crystal structure is stabilized by intermolecular weak interactions, for example, C–H···O, C–H···N, and C–H···S hydrogen bonds. Besides, the photoluminescence of 1 has been investigated. This compound exhibits photoluminescence with an emission maximum of 321 nm upon excitation at 259 nm. The present work is a combined experimental and computational study. Quantum chemical parameters such as bond lengths, bond angles, HOMO–LUMO energy levels, energy band gap \(\Delta E\), chemical hardness η, the dipole moment μ, and Natural bond orbital (NBO) analysis of the compound were investigated using the DFT at M06/GENECP (6-31 g(d) and LANL2DZ) basis sets for compound 1. Our calculation results have shown that compound 1 has an energy band gap (∆E = 3.857 eV), indicating a high recommendation for a semiconductor compound. The optimized geometry of the Cd(II) coordination polymer is shown in good agreement with single crystal X-ray data. UV/Vis spectra are calculated with the time-dependent density functional theory (TD-DFT) method. Furthermore, TD-DFT displays intraligand transition (ILCT) between triazole bridging ligands. Moreover, NBO analysis of the Cd(II) coordination polymer indicates that the lone pair donor orbital interaction between the N, S, and O lone pairs and π*, σ* anti-bonding orbitals provides stronger stability. Additionally, Solid-state density functional theory (DFT) calculations were performed on 1 using the program VASP to understand the electronic states and conduction pathways of the coordination polymer.
期刊介绍:
Transition Metal Chemistry is an international journal designed to deal with all aspects of the subject embodied in the title: the preparation of transition metal-based molecular compounds of all kinds (including complexes of the Group 12 elements), their structural, physical, kinetic, catalytic and biological properties, their use in chemical synthesis as well as their application in the widest context, their role in naturally occurring systems etc.
Manuscripts submitted to the journal should be of broad appeal to the readership and for this reason, papers which are confined to more specialised studies such as the measurement of solution phase equilibria or thermal decomposition studies, or papers which include extensive material on f-block elements, or papers dealing with non-molecular materials, will not normally be considered for publication. Work describing new ligands or coordination geometries must provide sufficient evidence for the confident assignment of structural formulae; this will usually take the form of one or more X-ray crystal structures.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
