Mustapha Abdullahi , Adamu Uzairu , Gideon Adamu Shallangwa , Paul Andrew Mamza , Muhammad Tukur Ibrahim
{"title":"通过遗传功能近似、比较分子场、分子对接和 ADMET/药代动力学研究,建立新型 Bornoel 类似物作为甲型流感病毒抑制剂的模型","authors":"Mustapha Abdullahi , Adamu Uzairu , Gideon Adamu Shallangwa , Paul Andrew Mamza , Muhammad Tukur Ibrahim","doi":"10.1016/j.ipha.2023.11.004","DOIUrl":null,"url":null,"abstract":"<div><p>Influenza A Virus (IAV) is a human respiratory pathogen prone to mutations and genome re-assortment leading to global pandemics. In this study, we applied the molecular modelling strategies such as, two-dimensional (2D), three-dimensional (3D)-quantitative structure–activity relationship (QSAR), and molecular docking simulation on a novel series of borneol compounds as influenza inhibitors. The best developed 2D-QSAR models, MLR (Q<sup>2</sup> = 0.8735, R<sup>2</sup> <sub>(train)</sub> = 0.9096) and ANN [3-2-1] (Q<sup>2</sup> = 0.8987, R<sup>2</sup><sub>(train)</sub> = 0.9171) revealed good and acceptable statistical validation metrics for the inhibitory activity predictions. The 3D-QSAR models were generated using the comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA), which showed CoMFA_S + E (Q<sup>2</sup> = 0.559, R<sup>2</sup><sub>(train)</sub> = 0.939) and CoMSIA_S + E (Q<sup>2</sup> = 0.577, R<sup>2</sup><sub>(train)</sub> = 0.941) as the best-observed models in accordance with the model acceptability standards. In addition, the contour maps generated from the CoMFA and CoMSIA models illustrates the steric and electrostatic molecular field relationships with the inhibitory effects of the studied molecules. Moreover, the binding modes of the active ligands were studied through molecular docking simulation with the Human Hemagglutinin (HA) receptor of influenza A virus (A/Puerto Rico/8/34(H1N1)). The studied compounds revealed the formation of H-bonds, CH-bonds, and hydrophobic interactions with the active amino acid residues such as Asn 543, Asn 614, Asn 617, Leu 618, Ser 540, Lys 539, and Lys 621 in the HA binding cavity. The prediction of drug-likeness and ADMET properties of the compounds revealed their good bioavailability and pharmacokinetic profiling. This study may provide a valuable <em>in-silico</em> guideline for discovering novel potent influenza inhibitors.</p></div>","PeriodicalId":100682,"journal":{"name":"Intelligent Pharmacy","volume":"2 2","pages":"Pages 190-203"},"PeriodicalIF":0.0000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2949866X23001181/pdfft?md5=a24a770f820d6cc9021c6b00a3d015cc&pid=1-s2.0-S2949866X23001181-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Modelling of novel bornoel analogs as Influenza A Virus inhibitors through genetic function approximation, comparative molecular fields, molecular docking, and ADMET/Pharmacokinetic studies\",\"authors\":\"Mustapha Abdullahi , Adamu Uzairu , Gideon Adamu Shallangwa , Paul Andrew Mamza , Muhammad Tukur Ibrahim\",\"doi\":\"10.1016/j.ipha.2023.11.004\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Influenza A Virus (IAV) is a human respiratory pathogen prone to mutations and genome re-assortment leading to global pandemics. In this study, we applied the molecular modelling strategies such as, two-dimensional (2D), three-dimensional (3D)-quantitative structure–activity relationship (QSAR), and molecular docking simulation on a novel series of borneol compounds as influenza inhibitors. The best developed 2D-QSAR models, MLR (Q<sup>2</sup> = 0.8735, R<sup>2</sup> <sub>(train)</sub> = 0.9096) and ANN [3-2-1] (Q<sup>2</sup> = 0.8987, R<sup>2</sup><sub>(train)</sub> = 0.9171) revealed good and acceptable statistical validation metrics for the inhibitory activity predictions. The 3D-QSAR models were generated using the comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA), which showed CoMFA_S + E (Q<sup>2</sup> = 0.559, R<sup>2</sup><sub>(train)</sub> = 0.939) and CoMSIA_S + E (Q<sup>2</sup> = 0.577, R<sup>2</sup><sub>(train)</sub> = 0.941) as the best-observed models in accordance with the model acceptability standards. In addition, the contour maps generated from the CoMFA and CoMSIA models illustrates the steric and electrostatic molecular field relationships with the inhibitory effects of the studied molecules. Moreover, the binding modes of the active ligands were studied through molecular docking simulation with the Human Hemagglutinin (HA) receptor of influenza A virus (A/Puerto Rico/8/34(H1N1)). The studied compounds revealed the formation of H-bonds, CH-bonds, and hydrophobic interactions with the active amino acid residues such as Asn 543, Asn 614, Asn 617, Leu 618, Ser 540, Lys 539, and Lys 621 in the HA binding cavity. The prediction of drug-likeness and ADMET properties of the compounds revealed their good bioavailability and pharmacokinetic profiling. This study may provide a valuable <em>in-silico</em> guideline for discovering novel potent influenza inhibitors.</p></div>\",\"PeriodicalId\":100682,\"journal\":{\"name\":\"Intelligent Pharmacy\",\"volume\":\"2 2\",\"pages\":\"Pages 190-203\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2949866X23001181/pdfft?md5=a24a770f820d6cc9021c6b00a3d015cc&pid=1-s2.0-S2949866X23001181-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Intelligent Pharmacy\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2949866X23001181\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/11/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Intelligent Pharmacy","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2949866X23001181","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/11/22 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Modelling of novel bornoel analogs as Influenza A Virus inhibitors through genetic function approximation, comparative molecular fields, molecular docking, and ADMET/Pharmacokinetic studies
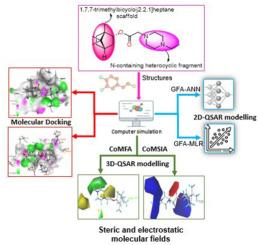
Influenza A Virus (IAV) is a human respiratory pathogen prone to mutations and genome re-assortment leading to global pandemics. In this study, we applied the molecular modelling strategies such as, two-dimensional (2D), three-dimensional (3D)-quantitative structure–activity relationship (QSAR), and molecular docking simulation on a novel series of borneol compounds as influenza inhibitors. The best developed 2D-QSAR models, MLR (Q2 = 0.8735, R2(train) = 0.9096) and ANN [3-2-1] (Q2 = 0.8987, R2(train) = 0.9171) revealed good and acceptable statistical validation metrics for the inhibitory activity predictions. The 3D-QSAR models were generated using the comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA), which showed CoMFA_S + E (Q2 = 0.559, R2(train) = 0.939) and CoMSIA_S + E (Q2 = 0.577, R2(train) = 0.941) as the best-observed models in accordance with the model acceptability standards. In addition, the contour maps generated from the CoMFA and CoMSIA models illustrates the steric and electrostatic molecular field relationships with the inhibitory effects of the studied molecules. Moreover, the binding modes of the active ligands were studied through molecular docking simulation with the Human Hemagglutinin (HA) receptor of influenza A virus (A/Puerto Rico/8/34(H1N1)). The studied compounds revealed the formation of H-bonds, CH-bonds, and hydrophobic interactions with the active amino acid residues such as Asn 543, Asn 614, Asn 617, Leu 618, Ser 540, Lys 539, and Lys 621 in the HA binding cavity. The prediction of drug-likeness and ADMET properties of the compounds revealed their good bioavailability and pharmacokinetic profiling. This study may provide a valuable in-silico guideline for discovering novel potent influenza inhibitors.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
