{"title":"利用多目标进化算法预测蛋白质的多重构象","authors":"Minghua Hou, Sirong Jin, Xinyue Cui, Chunxiang Peng, Kailong Zhao, Le Song, Guijun Zhang","doi":"10.1007/s12539-023-00597-5","DOIUrl":null,"url":null,"abstract":"<p><p>The breakthrough of AlphaFold2 and the publication of AlphaFold DB represent a significant advance in the field of predicting static protein structures. However, AlphaFold2 models tend to represent a single static structure, and multiple-conformation prediction remains a challenge. In this work, we proposed a method named MultiSFold, which uses a distance-based multi-objective evolutionary algorithm to predict multiple conformations. To begin, multiple energy landscapes are constructed using different competing constraints generated by deep learning. Subsequently, an iterative modal exploration and exploitation strategy is designed to sample conformations, incorporating multi-objective optimization, geometric optimization and structural similarity clustering. Finally, the final population is generated using a loop-specific sampling strategy to adjust the spatial orientations. MultiSFold was evaluated against state-of-the-art methods using a benchmark set containing 80 protein targets, each characterized by two representative conformational states. Based on the proposed metric, MultiSFold achieves a remarkable success ratio of 56.25% in predicting multiple conformations, while AlphaFold2 only achieves 10.00%, which may indicate that conformational sampling combined with knowledge gained through deep learning has the potential to generate conformations spanning the range between different conformational states. In addition, MultiSFold was tested on 244 human proteins with low structural accuracy in AlphaFold DB to test whether it could further improve the accuracy of static structures. The experimental results demonstrate the performance of MultiSFold, with a TM-score better than that of AlphaFold2 by 2.97% and RoseTTAFold by 7.72%. The online server is at http://zhanglab-bioinf.com/MultiSFold .</p>","PeriodicalId":13670,"journal":{"name":"Interdisciplinary Sciences: Computational Life Sciences","volume":" ","pages":"519-531"},"PeriodicalIF":3.9000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Protein Multiple Conformation Prediction Using Multi-Objective Evolution Algorithm.\",\"authors\":\"Minghua Hou, Sirong Jin, Xinyue Cui, Chunxiang Peng, Kailong Zhao, Le Song, Guijun Zhang\",\"doi\":\"10.1007/s12539-023-00597-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The breakthrough of AlphaFold2 and the publication of AlphaFold DB represent a significant advance in the field of predicting static protein structures. However, AlphaFold2 models tend to represent a single static structure, and multiple-conformation prediction remains a challenge. In this work, we proposed a method named MultiSFold, which uses a distance-based multi-objective evolutionary algorithm to predict multiple conformations. To begin, multiple energy landscapes are constructed using different competing constraints generated by deep learning. Subsequently, an iterative modal exploration and exploitation strategy is designed to sample conformations, incorporating multi-objective optimization, geometric optimization and structural similarity clustering. Finally, the final population is generated using a loop-specific sampling strategy to adjust the spatial orientations. MultiSFold was evaluated against state-of-the-art methods using a benchmark set containing 80 protein targets, each characterized by two representative conformational states. Based on the proposed metric, MultiSFold achieves a remarkable success ratio of 56.25% in predicting multiple conformations, while AlphaFold2 only achieves 10.00%, which may indicate that conformational sampling combined with knowledge gained through deep learning has the potential to generate conformations spanning the range between different conformational states. In addition, MultiSFold was tested on 244 human proteins with low structural accuracy in AlphaFold DB to test whether it could further improve the accuracy of static structures. The experimental results demonstrate the performance of MultiSFold, with a TM-score better than that of AlphaFold2 by 2.97% and RoseTTAFold by 7.72%. The online server is at http://zhanglab-bioinf.com/MultiSFold .</p>\",\"PeriodicalId\":13670,\"journal\":{\"name\":\"Interdisciplinary Sciences: Computational Life Sciences\",\"volume\":\" \",\"pages\":\"519-531\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2024-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Interdisciplinary Sciences: Computational Life Sciences\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s12539-023-00597-5\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/8 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Interdisciplinary Sciences: Computational Life Sciences","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s12539-023-00597-5","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/8 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Protein Multiple Conformation Prediction Using Multi-Objective Evolution Algorithm.
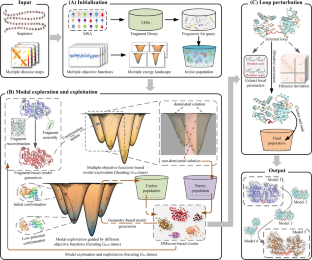
The breakthrough of AlphaFold2 and the publication of AlphaFold DB represent a significant advance in the field of predicting static protein structures. However, AlphaFold2 models tend to represent a single static structure, and multiple-conformation prediction remains a challenge. In this work, we proposed a method named MultiSFold, which uses a distance-based multi-objective evolutionary algorithm to predict multiple conformations. To begin, multiple energy landscapes are constructed using different competing constraints generated by deep learning. Subsequently, an iterative modal exploration and exploitation strategy is designed to sample conformations, incorporating multi-objective optimization, geometric optimization and structural similarity clustering. Finally, the final population is generated using a loop-specific sampling strategy to adjust the spatial orientations. MultiSFold was evaluated against state-of-the-art methods using a benchmark set containing 80 protein targets, each characterized by two representative conformational states. Based on the proposed metric, MultiSFold achieves a remarkable success ratio of 56.25% in predicting multiple conformations, while AlphaFold2 only achieves 10.00%, which may indicate that conformational sampling combined with knowledge gained through deep learning has the potential to generate conformations spanning the range between different conformational states. In addition, MultiSFold was tested on 244 human proteins with low structural accuracy in AlphaFold DB to test whether it could further improve the accuracy of static structures. The experimental results demonstrate the performance of MultiSFold, with a TM-score better than that of AlphaFold2 by 2.97% and RoseTTAFold by 7.72%. The online server is at http://zhanglab-bioinf.com/MultiSFold .
期刊介绍:
Interdisciplinary Sciences--Computational Life Sciences aims to cover the most recent and outstanding developments in interdisciplinary areas of sciences, especially focusing on computational life sciences, an area that is enjoying rapid development at the forefront of scientific research and technology.
The journal publishes original papers of significant general interest covering recent research and developments. Articles will be published rapidly by taking full advantage of internet technology for online submission and peer-reviewing of manuscripts, and then by publishing OnlineFirstTM through SpringerLink even before the issue is built or sent to the printer.
The editorial board consists of many leading scientists with international reputation, among others, Luc Montagnier (UNESCO, France), Dennis Salahub (University of Calgary, Canada), Weitao Yang (Duke University, USA). Prof. Dongqing Wei at the Shanghai Jiatong University is appointed as the editor-in-chief; he made important contributions in bioinformatics and computational physics and is best known for his ground-breaking works on the theory of ferroelectric liquids. With the help from a team of associate editors and the editorial board, an international journal with sound reputation shall be created.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
