{"title":"确定乙型肝炎病毒所致肝纤维化的新诊断靶标","authors":"Ying Wang, Shuo Qin, Meng Yang, Xiaoling Wang","doi":"10.1002/ila2.30","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Background</h3>\n \n <p>Liver fibrosis is a transitional stage from hepatitis to cirrhosis, and hepatitis B virus (HBV) is the most common cause of liver disease. Transcriptome sequencing technology and bioinformatics analysis are increasingly being used to screen diagnostic targets for liver fibrosis.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>The GSE171294 dataset of HBV-induced liver fibrosis tissue and normal tissue was obtained from the Gene Expression Omnibus (GEO) public database and used to screen for differentially expressed mRNAs using R software. mRNAs with |log fold change| >1 and <i>p</i> < 0.05 were considered to be differentially expressed. A heat map was drawn to visualize the expression patterns of the differentially expressed mRNAs. To screen for candidate target mRNAs, the differentially expressed mRNAs were annotated by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis. Finally, a protein–protein interaction (PPI) network was constructed to analyze the relationships between the differentially expressed mRNAs.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>A total of 243 differentially expressed mRNAs were identified (<i>p</i> < 0.05); 129 were up-regulated and 114 were down-regulated. The up-regulated and down-regulated mRNAs were significantly enriched in 16 and 8 KEGG pathways, respectively. The enriched KEGG pathways included Salmonella infection, Protein processing in the endoplasmic reticulum, IL-17 signaling pathway, and Aldosterone synthesis and secretion. The enriched GO terms were related mainly to cell proliferation, apoptosis, endoplasmic reticulum complex assembly, and myosin synthesis. The PPI network contained 161 nodes and 120 pairs of interactions. The top 10 key nodes were <i>CAV1, CD4, NR3C1, PDIA3, EZR, IRF4, SOX9, HSP90AB1, CD40,</i> and <i>SEC13</i>.</p>\n </section>\n \n <section>\n \n <h3> Conclusions</h3>\n \n <p>Bioinformatics analysis of the transcriptome sequencing data in the GSE171294 dataset identified <i>CD4</i>, <i>NR3C1</i>, and <i>EZR</i> and other genes at key nodes as new targets for the treatment of liver fibrosis caused by HBV. These results provide new insights for HBV-induced liver fibrosis research and clinical treatment.</p>\n </section>\n </div>","PeriodicalId":100656,"journal":{"name":"iLABMED","volume":"2 1","pages":"27-37"},"PeriodicalIF":0.0000,"publicationDate":"2024-01-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ila2.30","citationCount":"0","resultStr":"{\"title\":\"Identification of new diagnostic targets for hepatitis B virus-induced liver fibrosis\",\"authors\":\"Ying Wang, Shuo Qin, Meng Yang, Xiaoling Wang\",\"doi\":\"10.1002/ila2.30\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Background</h3>\\n \\n <p>Liver fibrosis is a transitional stage from hepatitis to cirrhosis, and hepatitis B virus (HBV) is the most common cause of liver disease. Transcriptome sequencing technology and bioinformatics analysis are increasingly being used to screen diagnostic targets for liver fibrosis.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>The GSE171294 dataset of HBV-induced liver fibrosis tissue and normal tissue was obtained from the Gene Expression Omnibus (GEO) public database and used to screen for differentially expressed mRNAs using R software. mRNAs with |log fold change| >1 and <i>p</i> < 0.05 were considered to be differentially expressed. A heat map was drawn to visualize the expression patterns of the differentially expressed mRNAs. To screen for candidate target mRNAs, the differentially expressed mRNAs were annotated by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis. Finally, a protein–protein interaction (PPI) network was constructed to analyze the relationships between the differentially expressed mRNAs.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>A total of 243 differentially expressed mRNAs were identified (<i>p</i> < 0.05); 129 were up-regulated and 114 were down-regulated. The up-regulated and down-regulated mRNAs were significantly enriched in 16 and 8 KEGG pathways, respectively. The enriched KEGG pathways included Salmonella infection, Protein processing in the endoplasmic reticulum, IL-17 signaling pathway, and Aldosterone synthesis and secretion. The enriched GO terms were related mainly to cell proliferation, apoptosis, endoplasmic reticulum complex assembly, and myosin synthesis. The PPI network contained 161 nodes and 120 pairs of interactions. The top 10 key nodes were <i>CAV1, CD4, NR3C1, PDIA3, EZR, IRF4, SOX9, HSP90AB1, CD40,</i> and <i>SEC13</i>.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusions</h3>\\n \\n <p>Bioinformatics analysis of the transcriptome sequencing data in the GSE171294 dataset identified <i>CD4</i>, <i>NR3C1</i>, and <i>EZR</i> and other genes at key nodes as new targets for the treatment of liver fibrosis caused by HBV. These results provide new insights for HBV-induced liver fibrosis research and clinical treatment.</p>\\n </section>\\n </div>\",\"PeriodicalId\":100656,\"journal\":{\"name\":\"iLABMED\",\"volume\":\"2 1\",\"pages\":\"27-37\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-01-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ila2.30\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"iLABMED\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ila2.30\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"iLABMED","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ila2.30","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Identification of new diagnostic targets for hepatitis B virus-induced liver fibrosis
Background
Liver fibrosis is a transitional stage from hepatitis to cirrhosis, and hepatitis B virus (HBV) is the most common cause of liver disease. Transcriptome sequencing technology and bioinformatics analysis are increasingly being used to screen diagnostic targets for liver fibrosis.
Methods
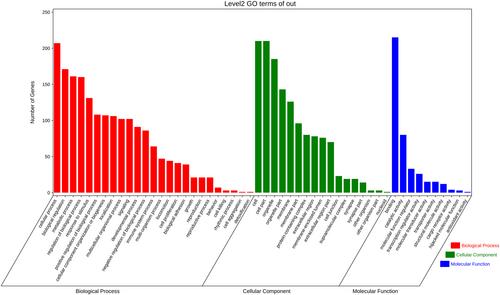
The GSE171294 dataset of HBV-induced liver fibrosis tissue and normal tissue was obtained from the Gene Expression Omnibus (GEO) public database and used to screen for differentially expressed mRNAs using R software. mRNAs with |log fold change| >1 and p < 0.05 were considered to be differentially expressed. A heat map was drawn to visualize the expression patterns of the differentially expressed mRNAs. To screen for candidate target mRNAs, the differentially expressed mRNAs were annotated by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis. Finally, a protein–protein interaction (PPI) network was constructed to analyze the relationships between the differentially expressed mRNAs.
Results
A total of 243 differentially expressed mRNAs were identified (p < 0.05); 129 were up-regulated and 114 were down-regulated. The up-regulated and down-regulated mRNAs were significantly enriched in 16 and 8 KEGG pathways, respectively. The enriched KEGG pathways included Salmonella infection, Protein processing in the endoplasmic reticulum, IL-17 signaling pathway, and Aldosterone synthesis and secretion. The enriched GO terms were related mainly to cell proliferation, apoptosis, endoplasmic reticulum complex assembly, and myosin synthesis. The PPI network contained 161 nodes and 120 pairs of interactions. The top 10 key nodes were CAV1, CD4, NR3C1, PDIA3, EZR, IRF4, SOX9, HSP90AB1, CD40, and SEC13.
Conclusions
Bioinformatics analysis of the transcriptome sequencing data in the GSE171294 dataset identified CD4, NR3C1, and EZR and other genes at key nodes as new targets for the treatment of liver fibrosis caused by HBV. These results provide new insights for HBV-induced liver fibrosis research and clinical treatment.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
