Giulio Tesei, Anna Ida Trolle, Nicolas Jonsson, Johannes Betz, Frederik E. Knudsen, Francesco Pesce, Kristoffer E. Johansson, Kresten Lindorff-Larsen
{"title":"人类内在无序蛋白质组的构象组合","authors":"Giulio Tesei, Anna Ida Trolle, Nicolas Jonsson, Johannes Betz, Frederik E. Knudsen, Francesco Pesce, Kristoffer E. Johansson, Kresten Lindorff-Larsen","doi":"10.1038/s41586-023-07004-5","DOIUrl":null,"url":null,"abstract":"Intrinsically disordered proteins and regions (collectively, IDRs) are pervasive across proteomes in all kingdoms of life, help to shape biological functions and are involved in numerous diseases. IDRs populate a diverse set of transiently formed structures and defy conventional sequence–structure–function relationships1. Developments in protein science have made it possible to predict the three-dimensional structures of folded proteins at the proteome scale2. By contrast, there is a lack of knowledge about the conformational properties of IDRs, partly because the sequences of disordered proteins are poorly conserved and also because only a few of these proteins have been characterized experimentally. The inability to predict structural properties of IDRs across the proteome has limited our understanding of the functional roles of IDRs and how evolution shapes them. As a supplement to previous structural studies of individual IDRs3, we developed an efficient molecular model to generate conformational ensembles of IDRs and thereby to predict their conformational properties from sequences4,5. Here we use this model to simulate nearly all of the IDRs in the human proteome. Examining conformational ensembles of 28,058 IDRs, we show how chain compaction is correlated with cellular function and localization. We provide insights into how sequence features relate to chain compaction and, using a machine-learning model trained on our simulation data, show the conservation of conformational properties across orthologues. Our results recapitulate observations from previous studies of individual protein systems and exemplify how to link—at the proteome scale—conformational ensembles with cellular function and localization, amino acid sequence, evolutionary conservation and disease variants. Our freely available database of conformational properties will encourage further experimental investigation and enable the generation of hypotheses about the biological roles and evolution of IDRs. A computational model generates conformational ensembles of 28,058 intrinsically disordered proteins and regions (IDRs) in the human proteome and sheds light on the relationship between sequence, conformational properties and functions of IDRs.","PeriodicalId":18787,"journal":{"name":"Nature","volume":"626 8000","pages":"897-904"},"PeriodicalIF":48.5000,"publicationDate":"2024-01-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Conformational ensembles of the human intrinsically disordered proteome\",\"authors\":\"Giulio Tesei, Anna Ida Trolle, Nicolas Jonsson, Johannes Betz, Frederik E. Knudsen, Francesco Pesce, Kristoffer E. Johansson, Kresten Lindorff-Larsen\",\"doi\":\"10.1038/s41586-023-07004-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Intrinsically disordered proteins and regions (collectively, IDRs) are pervasive across proteomes in all kingdoms of life, help to shape biological functions and are involved in numerous diseases. IDRs populate a diverse set of transiently formed structures and defy conventional sequence–structure–function relationships1. Developments in protein science have made it possible to predict the three-dimensional structures of folded proteins at the proteome scale2. By contrast, there is a lack of knowledge about the conformational properties of IDRs, partly because the sequences of disordered proteins are poorly conserved and also because only a few of these proteins have been characterized experimentally. The inability to predict structural properties of IDRs across the proteome has limited our understanding of the functional roles of IDRs and how evolution shapes them. As a supplement to previous structural studies of individual IDRs3, we developed an efficient molecular model to generate conformational ensembles of IDRs and thereby to predict their conformational properties from sequences4,5. Here we use this model to simulate nearly all of the IDRs in the human proteome. Examining conformational ensembles of 28,058 IDRs, we show how chain compaction is correlated with cellular function and localization. We provide insights into how sequence features relate to chain compaction and, using a machine-learning model trained on our simulation data, show the conservation of conformational properties across orthologues. Our results recapitulate observations from previous studies of individual protein systems and exemplify how to link—at the proteome scale—conformational ensembles with cellular function and localization, amino acid sequence, evolutionary conservation and disease variants. Our freely available database of conformational properties will encourage further experimental investigation and enable the generation of hypotheses about the biological roles and evolution of IDRs. A computational model generates conformational ensembles of 28,058 intrinsically disordered proteins and regions (IDRs) in the human proteome and sheds light on the relationship between sequence, conformational properties and functions of IDRs.\",\"PeriodicalId\":18787,\"journal\":{\"name\":\"Nature\",\"volume\":\"626 8000\",\"pages\":\"897-904\"},\"PeriodicalIF\":48.5000,\"publicationDate\":\"2024-01-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature\",\"FirstCategoryId\":\"103\",\"ListUrlMain\":\"https://www.nature.com/articles/s41586-023-07004-5\",\"RegionNum\":1,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature","FirstCategoryId":"103","ListUrlMain":"https://www.nature.com/articles/s41586-023-07004-5","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
Conformational ensembles of the human intrinsically disordered proteome
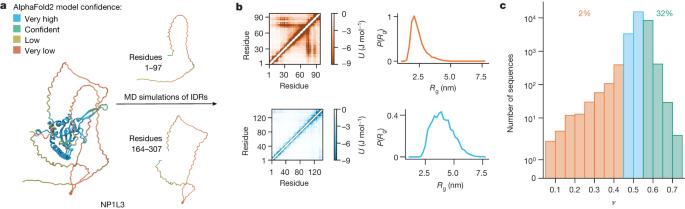
Intrinsically disordered proteins and regions (collectively, IDRs) are pervasive across proteomes in all kingdoms of life, help to shape biological functions and are involved in numerous diseases. IDRs populate a diverse set of transiently formed structures and defy conventional sequence–structure–function relationships1. Developments in protein science have made it possible to predict the three-dimensional structures of folded proteins at the proteome scale2. By contrast, there is a lack of knowledge about the conformational properties of IDRs, partly because the sequences of disordered proteins are poorly conserved and also because only a few of these proteins have been characterized experimentally. The inability to predict structural properties of IDRs across the proteome has limited our understanding of the functional roles of IDRs and how evolution shapes them. As a supplement to previous structural studies of individual IDRs3, we developed an efficient molecular model to generate conformational ensembles of IDRs and thereby to predict their conformational properties from sequences4,5. Here we use this model to simulate nearly all of the IDRs in the human proteome. Examining conformational ensembles of 28,058 IDRs, we show how chain compaction is correlated with cellular function and localization. We provide insights into how sequence features relate to chain compaction and, using a machine-learning model trained on our simulation data, show the conservation of conformational properties across orthologues. Our results recapitulate observations from previous studies of individual protein systems and exemplify how to link—at the proteome scale—conformational ensembles with cellular function and localization, amino acid sequence, evolutionary conservation and disease variants. Our freely available database of conformational properties will encourage further experimental investigation and enable the generation of hypotheses about the biological roles and evolution of IDRs. A computational model generates conformational ensembles of 28,058 intrinsically disordered proteins and regions (IDRs) in the human proteome and sheds light on the relationship between sequence, conformational properties and functions of IDRs.
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
