{"title":"MetaLo:从分子相互作用图中提取的逻辑模型代谢分析。","authors":"Sahar Aghakhani, Anna Niarakis, Sylvain Soliman","doi":"10.1515/jib-2023-0048","DOIUrl":null,"url":null,"abstract":"<p><p>Molecular interaction maps (MIMs) are static graphical representations depicting complex biochemical networks that can be formalized using one of the Systems Biology Graphical Notation languages. Regardless of their extensive coverage of various biological processes, they are limited in terms of dynamic insights. However, MIMs can serve as templates for developing dynamic computational models. We present MetaLo, an open-source Python package that enables the coupling of Boolean models inferred from process description MIMs with generic core metabolic networks. MetaLo provides a framework to study the impact of signaling cascades, gene regulation processes, and metabolic flux distribution of central energy production pathways. MetaLo computes the Boolean model's asynchronous asymptotic behavior, through the identification of trap-spaces, and extracts metabolic constraints to contextualize the generic metabolic network. MetaLo is able to handle large-scale Boolean models and genome-scale metabolic models without requiring kinetic information or manual tuning. The framework behind MetaLo enables in depth analysis of the regulatory model, and may allow tackling a lack of omics data in poorly addressed biological fields to contextualize generic metabolic networks along with improper automatic reconstructions of cell- and/or disease-specific metabolic networks. MetaLo is available at https://pypi.org/project/metalo/ under the terms of the GNU General Public License v3.</p>","PeriodicalId":53625,"journal":{"name":"Journal of Integrative Bioinformatics","volume":" ","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2024-02-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11293895/pdf/","citationCount":"0","resultStr":"{\"title\":\"MetaLo: metabolic analysis of Logical models extracted from molecular interaction maps.\",\"authors\":\"Sahar Aghakhani, Anna Niarakis, Sylvain Soliman\",\"doi\":\"10.1515/jib-2023-0048\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Molecular interaction maps (MIMs) are static graphical representations depicting complex biochemical networks that can be formalized using one of the Systems Biology Graphical Notation languages. Regardless of their extensive coverage of various biological processes, they are limited in terms of dynamic insights. However, MIMs can serve as templates for developing dynamic computational models. We present MetaLo, an open-source Python package that enables the coupling of Boolean models inferred from process description MIMs with generic core metabolic networks. MetaLo provides a framework to study the impact of signaling cascades, gene regulation processes, and metabolic flux distribution of central energy production pathways. MetaLo computes the Boolean model's asynchronous asymptotic behavior, through the identification of trap-spaces, and extracts metabolic constraints to contextualize the generic metabolic network. MetaLo is able to handle large-scale Boolean models and genome-scale metabolic models without requiring kinetic information or manual tuning. The framework behind MetaLo enables in depth analysis of the regulatory model, and may allow tackling a lack of omics data in poorly addressed biological fields to contextualize generic metabolic networks along with improper automatic reconstructions of cell- and/or disease-specific metabolic networks. MetaLo is available at https://pypi.org/project/metalo/ under the terms of the GNU General Public License v3.</p>\",\"PeriodicalId\":53625,\"journal\":{\"name\":\"Journal of Integrative Bioinformatics\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-02-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11293895/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Integrative Bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1515/jib-2023-0048\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/3/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Integrative Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1515/jib-2023-0048","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
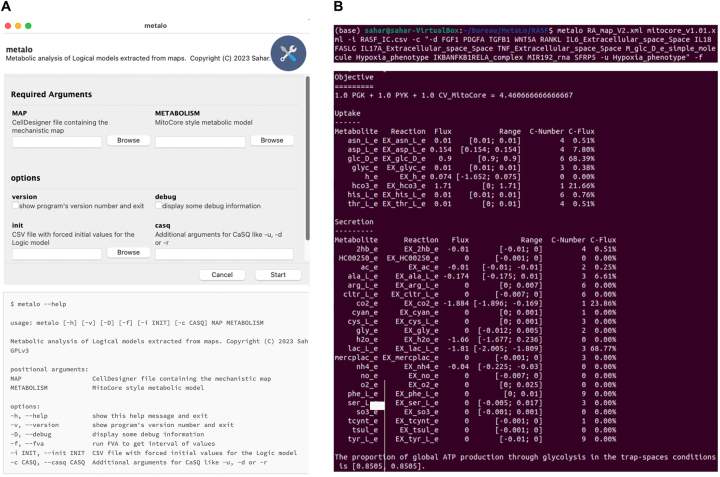
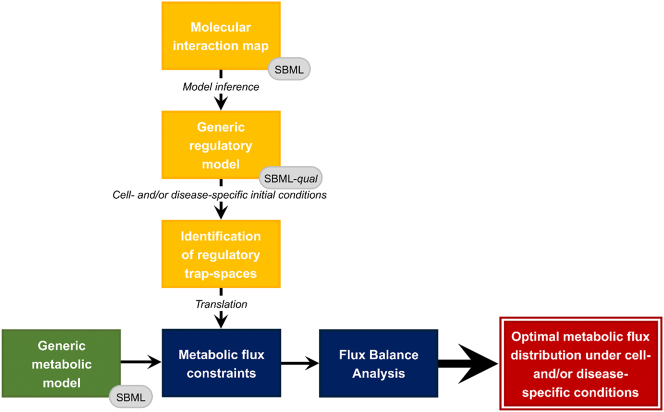
分子相互作用图(MIM)是描述复杂生化网络的静态图形表示法,可使用系统生物学图形符号语言之一进行形式化。尽管它们广泛覆盖了各种生物过程,但在动态洞察方面却很有限。然而,MIM 可以作为开发动态计算模型的模板。我们介绍的 MetaLo 是一个开源 Python 软件包,它能将从过程描述 MIMs 中推断出的布尔模型与通用核心代谢网络相耦合。MetaLo 提供了一个框架,用于研究信号级联、基因调控过程和中心能量生产途径的代谢通量分布的影响。MetaLo 通过识别陷阱空间来计算布尔模型的异步渐进行为,并提取代谢约束条件,从而将通用代谢网络背景化。MetaLo 能够处理大规模布尔模型和基因组规模的代谢模型,而无需动力学信息或人工调整。MetaLo 背后的框架可对调控模型进行深入分析,并可解决生物领域中缺乏 omics 数据的问题,从而将通用代谢网络与细胞和/或疾病特定代谢网络的不当自动重建结合起来。MetaLo 根据 GNU 通用公共许可证 v3 条款发布于 https://pypi.org/project/metalo/。
MetaLo: metabolic analysis of Logical models extracted from molecular interaction maps.
Molecular interaction maps (MIMs) are static graphical representations depicting complex biochemical networks that can be formalized using one of the Systems Biology Graphical Notation languages. Regardless of their extensive coverage of various biological processes, they are limited in terms of dynamic insights. However, MIMs can serve as templates for developing dynamic computational models. We present MetaLo, an open-source Python package that enables the coupling of Boolean models inferred from process description MIMs with generic core metabolic networks. MetaLo provides a framework to study the impact of signaling cascades, gene regulation processes, and metabolic flux distribution of central energy production pathways. MetaLo computes the Boolean model's asynchronous asymptotic behavior, through the identification of trap-spaces, and extracts metabolic constraints to contextualize the generic metabolic network. MetaLo is able to handle large-scale Boolean models and genome-scale metabolic models without requiring kinetic information or manual tuning. The framework behind MetaLo enables in depth analysis of the regulatory model, and may allow tackling a lack of omics data in poorly addressed biological fields to contextualize generic metabolic networks along with improper automatic reconstructions of cell- and/or disease-specific metabolic networks. MetaLo is available at https://pypi.org/project/metalo/ under the terms of the GNU General Public License v3.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
