Jia Wei, Junyi Yang, Linpo Yang, Yuanzuo Li, Yinglin Song
{"title":"分子结构修饰烯衍生物的可调三阶 NLO 性能","authors":"Jia Wei, Junyi Yang, Linpo Yang, Yuanzuo Li, Yinglin Song","doi":"10.1002/qua.27354","DOIUrl":null,"url":null,"abstract":"<p>The substitution of heteroatoms and the expansion of π-conjugated units have significant effects on the photoelectric properties of polycyclic aromatic hydrocarbons (PAHs). In this study, based on the experimental molecule PBC, 10 acene derivatives containing the pyrrole group were designed by three strategies: (1) changing the connection position of the pyrrole group, (2) exchanging the position of the N atom, and (3) increasing the length of the π-linker. Density functional theory (DFT) was used to optimize the molecular geometric structure. Time-dependent density functional theory (TD-DFT) was used to calculate the relevant parameters of the excited states. The results show that both of the pyrrole group and the N atom in the para-position, and the addition of the π-linker can reduce the energy gap, cause redshift of the linear absorption peak, and increase the two photon absorption (TPA) cross-section. The analyses of the charge density difference (CDD) and the charge-transfer matrix (CTM) proved that the electron transfer is mainly concentrated in the π-linker. Moreover, the participation of the multi-ring skeleton on both sides decreases gradually with the increase of the length of the π-linker. The length of the π-linker changes the dominant transition channel and intramolecular charge-transfer (ICT) characteristics of TPA, thus affecting the transition dipole moments and nonlinear absorption properties. The second hyperpolarizability of the designed molecule PBI5-p5 is also significantly superior to that of similar materials reported. It is expected that the above molecular design strategies and comprehensive analysis of nonlinear optical (NLO) properties could provide theoretical support for the research on acene derivatives.</p>","PeriodicalId":182,"journal":{"name":"International Journal of Quantum Chemistry","volume":"124 4","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-02-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Tunable third-order NLO properties of acene derivatives with molecular structural modification\",\"authors\":\"Jia Wei, Junyi Yang, Linpo Yang, Yuanzuo Li, Yinglin Song\",\"doi\":\"10.1002/qua.27354\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The substitution of heteroatoms and the expansion of π-conjugated units have significant effects on the photoelectric properties of polycyclic aromatic hydrocarbons (PAHs). In this study, based on the experimental molecule PBC, 10 acene derivatives containing the pyrrole group were designed by three strategies: (1) changing the connection position of the pyrrole group, (2) exchanging the position of the N atom, and (3) increasing the length of the π-linker. Density functional theory (DFT) was used to optimize the molecular geometric structure. Time-dependent density functional theory (TD-DFT) was used to calculate the relevant parameters of the excited states. The results show that both of the pyrrole group and the N atom in the para-position, and the addition of the π-linker can reduce the energy gap, cause redshift of the linear absorption peak, and increase the two photon absorption (TPA) cross-section. The analyses of the charge density difference (CDD) and the charge-transfer matrix (CTM) proved that the electron transfer is mainly concentrated in the π-linker. Moreover, the participation of the multi-ring skeleton on both sides decreases gradually with the increase of the length of the π-linker. The length of the π-linker changes the dominant transition channel and intramolecular charge-transfer (ICT) characteristics of TPA, thus affecting the transition dipole moments and nonlinear absorption properties. The second hyperpolarizability of the designed molecule PBI5-p5 is also significantly superior to that of similar materials reported. It is expected that the above molecular design strategies and comprehensive analysis of nonlinear optical (NLO) properties could provide theoretical support for the research on acene derivatives.</p>\",\"PeriodicalId\":182,\"journal\":{\"name\":\"International Journal of Quantum Chemistry\",\"volume\":\"124 4\",\"pages\":\"\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2024-02-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Quantum Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/qua.27354\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Quantum Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/qua.27354","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
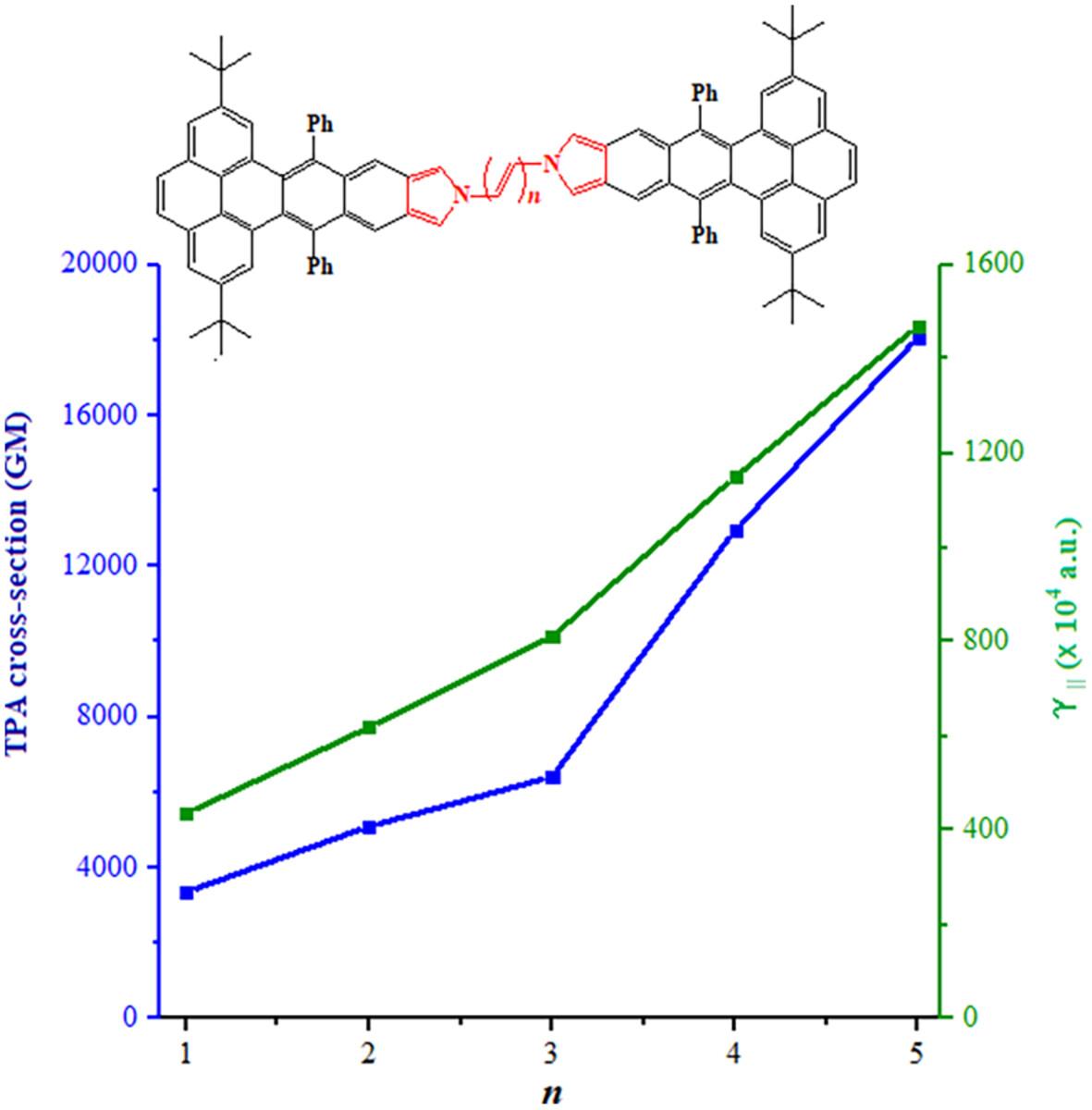
杂原子的取代和π-共轭单元的扩展对多环芳烃(PAHs)的光电特性有显著影响。本研究以实验分子 PBC 为基础,通过三种策略设计了 10 种含有吡咯基团的烯烃衍生物:(1) 改变吡咯基团的连接位置;(2) 交换 N 原子的位置;(3) 增加 π 连接物的长度。密度泛函理论(DFT)用于优化分子几何结构。随时间变化的密度泛函理论(TD-DFT)用于计算激发态的相关参数。结果表明,位于对位的吡咯基团和 N 原子以及添加的 π 连接剂都能减小能隙,导致线性吸收峰红移,并增加双光子吸收(TPA)截面。对电荷密度差(CDD)和电荷转移矩阵(CTM)的分析表明,电子转移主要集中在π-连接剂中。此外,随着π-连接体长度的增加,两侧多环骨架的参与程度逐渐降低。π-连接剂的长度改变了 TPA 的主要过渡通道和分子内电荷转移(ICT)特性,从而影响了过渡偶极矩和非线性吸收特性。所设计分子 PBI5-p5 的第二超极化性也明显优于已报道的同类材料。希望上述分子设计策略和非线性光学(NLO)特性的综合分析能为烯烃衍生物的研究提供理论支持。
Tunable third-order NLO properties of acene derivatives with molecular structural modification
The substitution of heteroatoms and the expansion of π-conjugated units have significant effects on the photoelectric properties of polycyclic aromatic hydrocarbons (PAHs). In this study, based on the experimental molecule PBC, 10 acene derivatives containing the pyrrole group were designed by three strategies: (1) changing the connection position of the pyrrole group, (2) exchanging the position of the N atom, and (3) increasing the length of the π-linker. Density functional theory (DFT) was used to optimize the molecular geometric structure. Time-dependent density functional theory (TD-DFT) was used to calculate the relevant parameters of the excited states. The results show that both of the pyrrole group and the N atom in the para-position, and the addition of the π-linker can reduce the energy gap, cause redshift of the linear absorption peak, and increase the two photon absorption (TPA) cross-section. The analyses of the charge density difference (CDD) and the charge-transfer matrix (CTM) proved that the electron transfer is mainly concentrated in the π-linker. Moreover, the participation of the multi-ring skeleton on both sides decreases gradually with the increase of the length of the π-linker. The length of the π-linker changes the dominant transition channel and intramolecular charge-transfer (ICT) characteristics of TPA, thus affecting the transition dipole moments and nonlinear absorption properties. The second hyperpolarizability of the designed molecule PBI5-p5 is also significantly superior to that of similar materials reported. It is expected that the above molecular design strategies and comprehensive analysis of nonlinear optical (NLO) properties could provide theoretical support for the research on acene derivatives.
期刊介绍:
Since its first formulation quantum chemistry has provided the conceptual and terminological framework necessary to understand atoms, molecules and the condensed matter. Over the past decades synergistic advances in the methodological developments, software and hardware have transformed quantum chemistry in a truly interdisciplinary science that has expanded beyond its traditional core of molecular sciences to fields as diverse as chemistry and catalysis, biophysics, nanotechnology and material science.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
