Hope A. Tanudisastro, Ira W. Deveson, Harriet Dashnow, Daniel G. MacArthur
{"title":"人类基因组中短串联重复序列的测序和特征描述。","authors":"Hope A. Tanudisastro, Ira W. Deveson, Harriet Dashnow, Daniel G. MacArthur","doi":"10.1038/s41576-024-00692-3","DOIUrl":null,"url":null,"abstract":"Short tandem repeats (STRs) are highly polymorphic sequences throughout the human genome that are composed of repeated copies of a 1–6-bp motif. Over 1 million variable STR loci are known, some of which regulate gene expression and influence complex traits, such as height. Moreover, variants in at least 60 STR loci cause genetic disorders, including Huntington disease and fragile X syndrome. Accurately identifying and genotyping STR variants is challenging, in particular mapping short reads to repetitive regions and inferring expanded repeat lengths. Recent advances in sequencing technology and computational tools for STR genotyping from sequencing data promise to help overcome this challenge and solve genetically unresolved cases and the ‘missing heritability’ of polygenic traits. Here, we compare STR genotyping methods, analytical tools and their applications to understand the effect of STR variation on health and disease. We identify emergent opportunities to refine genotyping and quality-control approaches as well as to integrate STRs into variant-calling workflows and large cohort analyses. This Review describes tools and approaches for characterizing short tandem repeats in the human genome from whole-genome sequencing data. Furthermore, the authors discuss how these recent developments have helped to better understand the effect of short tandem repeats on human health and disease.","PeriodicalId":19067,"journal":{"name":"Nature Reviews Genetics","volume":"25 7","pages":"460-475"},"PeriodicalIF":52.0000,"publicationDate":"2024-02-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Sequencing and characterizing short tandem repeats in the human genome\",\"authors\":\"Hope A. Tanudisastro, Ira W. Deveson, Harriet Dashnow, Daniel G. MacArthur\",\"doi\":\"10.1038/s41576-024-00692-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Short tandem repeats (STRs) are highly polymorphic sequences throughout the human genome that are composed of repeated copies of a 1–6-bp motif. Over 1 million variable STR loci are known, some of which regulate gene expression and influence complex traits, such as height. Moreover, variants in at least 60 STR loci cause genetic disorders, including Huntington disease and fragile X syndrome. Accurately identifying and genotyping STR variants is challenging, in particular mapping short reads to repetitive regions and inferring expanded repeat lengths. Recent advances in sequencing technology and computational tools for STR genotyping from sequencing data promise to help overcome this challenge and solve genetically unresolved cases and the ‘missing heritability’ of polygenic traits. Here, we compare STR genotyping methods, analytical tools and their applications to understand the effect of STR variation on health and disease. We identify emergent opportunities to refine genotyping and quality-control approaches as well as to integrate STRs into variant-calling workflows and large cohort analyses. This Review describes tools and approaches for characterizing short tandem repeats in the human genome from whole-genome sequencing data. Furthermore, the authors discuss how these recent developments have helped to better understand the effect of short tandem repeats on human health and disease.\",\"PeriodicalId\":19067,\"journal\":{\"name\":\"Nature Reviews Genetics\",\"volume\":\"25 7\",\"pages\":\"460-475\"},\"PeriodicalIF\":52.0000,\"publicationDate\":\"2024-02-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Reviews Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s41576-024-00692-3\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Reviews Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41576-024-00692-3","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Sequencing and characterizing short tandem repeats in the human genome
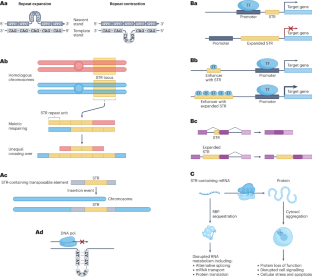
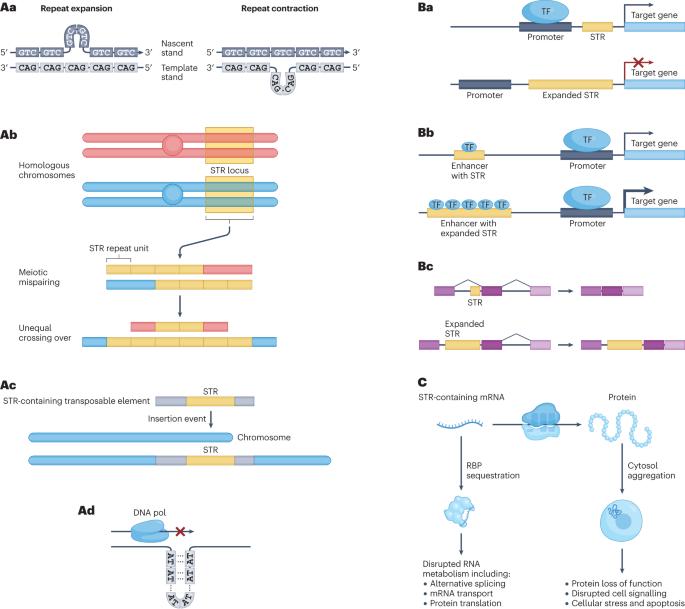
Short tandem repeats (STRs) are highly polymorphic sequences throughout the human genome that are composed of repeated copies of a 1–6-bp motif. Over 1 million variable STR loci are known, some of which regulate gene expression and influence complex traits, such as height. Moreover, variants in at least 60 STR loci cause genetic disorders, including Huntington disease and fragile X syndrome. Accurately identifying and genotyping STR variants is challenging, in particular mapping short reads to repetitive regions and inferring expanded repeat lengths. Recent advances in sequencing technology and computational tools for STR genotyping from sequencing data promise to help overcome this challenge and solve genetically unresolved cases and the ‘missing heritability’ of polygenic traits. Here, we compare STR genotyping methods, analytical tools and their applications to understand the effect of STR variation on health and disease. We identify emergent opportunities to refine genotyping and quality-control approaches as well as to integrate STRs into variant-calling workflows and large cohort analyses. This Review describes tools and approaches for characterizing short tandem repeats in the human genome from whole-genome sequencing data. Furthermore, the authors discuss how these recent developments have helped to better understand the effect of short tandem repeats on human health and disease.
期刊介绍:
At Nature Reviews Genetics, our goal is to be the leading source of reviews and commentaries for the scientific communities we serve. We are dedicated to publishing authoritative articles that are easily accessible to our readers. We believe in enhancing our articles with clear and understandable figures, tables, and other display items. Our aim is to provide an unparalleled service to authors, referees, and readers, and we are committed to maximizing the usefulness and impact of each article we publish.
Within our journal, we publish a range of content including Research Highlights, Comments, Reviews, and Perspectives that are relevant to geneticists and genomicists. With our broad scope, we ensure that the articles we publish reach the widest possible audience.
As part of the Nature Reviews portfolio of journals, we strive to uphold the high standards and reputation associated with this esteemed collection of publications.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
