{"title":"SAW:立体测序空间转录组学高效准确的数据分析工作流程。","authors":"Chun Gong, Shengkang Li, Leying Wang, Fuxiang Zhao, Shuangsang Fang, Dong Yuan, Zijian Zhao, Qiqi He, Mei Li, Weiqing Liu, Zhaoxun Li, Hongqing Xie, Sha Liao, Ao Chen, Yong Zhang, Yuxiang Li, Xun Xu","doi":"10.46471/gigabyte.111","DOIUrl":null,"url":null,"abstract":"<p><p>The basic analysis steps of spatial transcriptomics require obtaining gene expression information from both space and cells. The existing tools for these analyses incur performance issues when dealing with large datasets. These issues involve computationally intensive spatial localization, RNA genome alignment, and excessive memory usage in large chip scenarios. These problems affect the applicability and efficiency of the analysis. Here, a high-performance and accurate spatial transcriptomics data analysis workflow, called Stereo-seq Analysis Workflow (SAW), was developed for the Stereo-seq technology developed at BGI. SAW includes mRNA spatial position reconstruction, genome alignment, gene expression matrix generation, and clustering. The workflow outputs files in a universal format for subsequent personalized analysis. The execution time for the entire analysis is ∼148 min with 1 GB reads 1 × 1 cm chip test data, 1.8 times faster than with an unoptimized workflow.</p>","PeriodicalId":73157,"journal":{"name":"GigaByte (Hong Kong, China)","volume":"2024 ","pages":"gigabyte111"},"PeriodicalIF":1.2000,"publicationDate":"2024-02-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10905255/pdf/","citationCount":"0","resultStr":"{\"title\":\"SAW: an efficient and accurate data analysis workflow for Stereo-seq spatial transcriptomics.\",\"authors\":\"Chun Gong, Shengkang Li, Leying Wang, Fuxiang Zhao, Shuangsang Fang, Dong Yuan, Zijian Zhao, Qiqi He, Mei Li, Weiqing Liu, Zhaoxun Li, Hongqing Xie, Sha Liao, Ao Chen, Yong Zhang, Yuxiang Li, Xun Xu\",\"doi\":\"10.46471/gigabyte.111\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The basic analysis steps of spatial transcriptomics require obtaining gene expression information from both space and cells. The existing tools for these analyses incur performance issues when dealing with large datasets. These issues involve computationally intensive spatial localization, RNA genome alignment, and excessive memory usage in large chip scenarios. These problems affect the applicability and efficiency of the analysis. Here, a high-performance and accurate spatial transcriptomics data analysis workflow, called Stereo-seq Analysis Workflow (SAW), was developed for the Stereo-seq technology developed at BGI. SAW includes mRNA spatial position reconstruction, genome alignment, gene expression matrix generation, and clustering. The workflow outputs files in a universal format for subsequent personalized analysis. The execution time for the entire analysis is ∼148 min with 1 GB reads 1 × 1 cm chip test data, 1.8 times faster than with an unoptimized workflow.</p>\",\"PeriodicalId\":73157,\"journal\":{\"name\":\"GigaByte (Hong Kong, China)\",\"volume\":\"2024 \",\"pages\":\"gigabyte111\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2024-02-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10905255/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"GigaByte (Hong Kong, China)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.46471/gigabyte.111\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"GigaByte (Hong Kong, China)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.46471/gigabyte.111","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
SAW: an efficient and accurate data analysis workflow for Stereo-seq spatial transcriptomics.
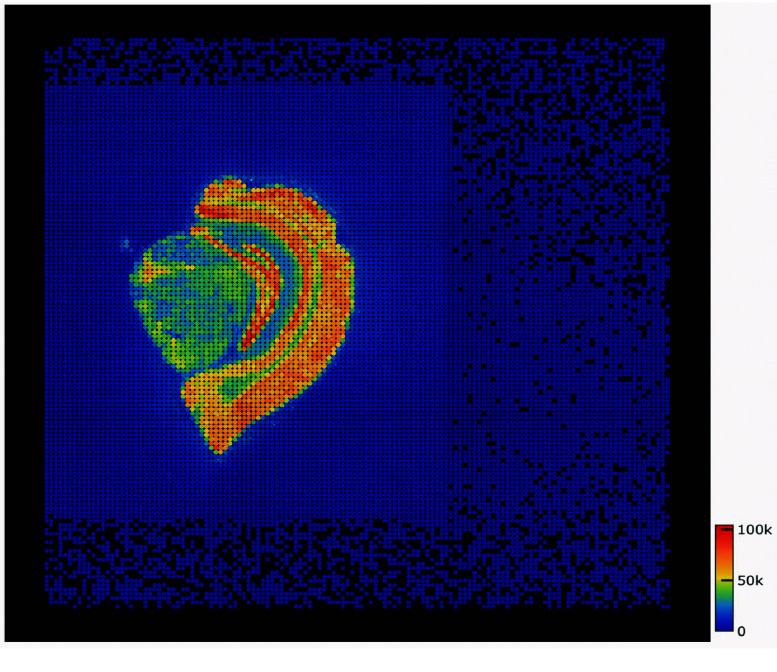
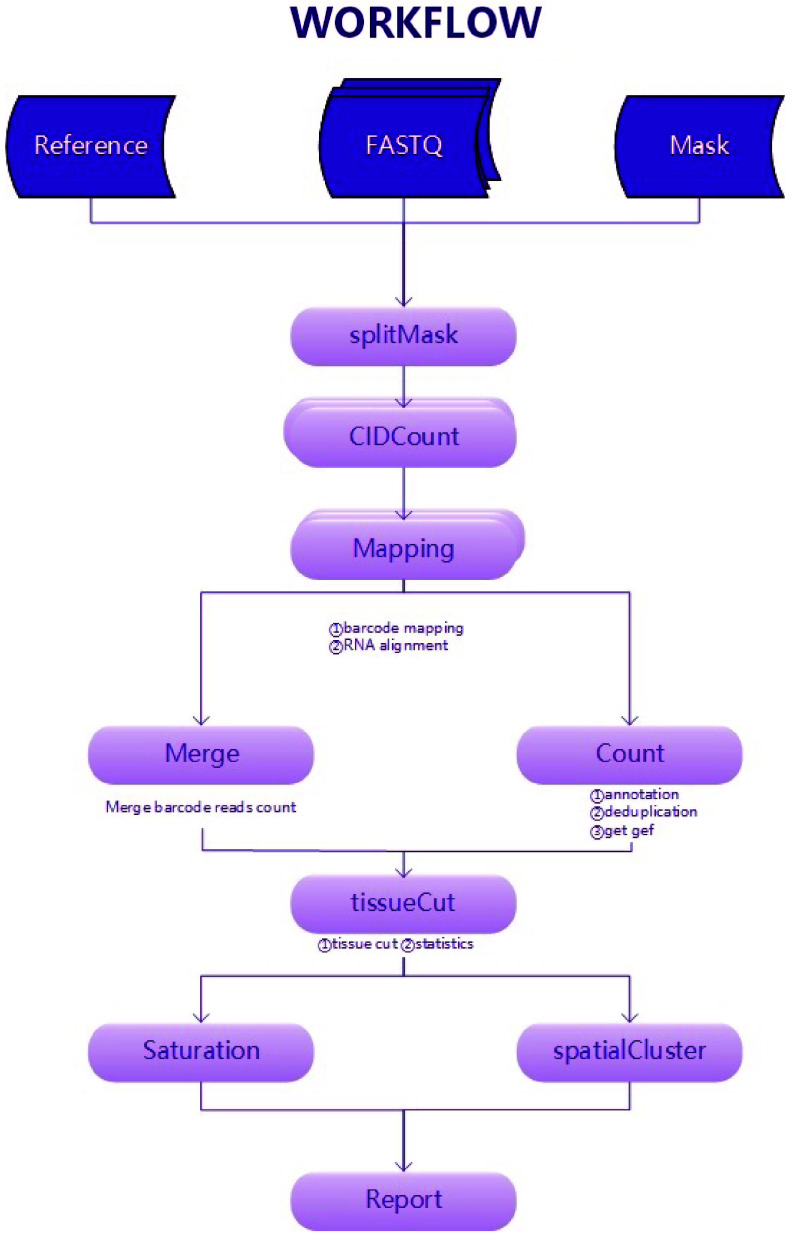
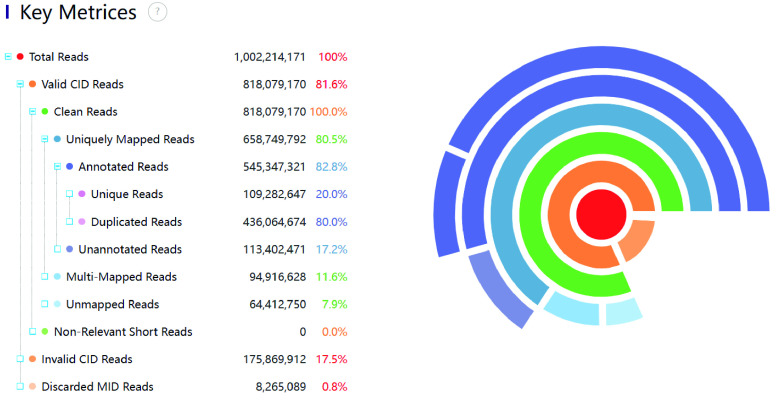
The basic analysis steps of spatial transcriptomics require obtaining gene expression information from both space and cells. The existing tools for these analyses incur performance issues when dealing with large datasets. These issues involve computationally intensive spatial localization, RNA genome alignment, and excessive memory usage in large chip scenarios. These problems affect the applicability and efficiency of the analysis. Here, a high-performance and accurate spatial transcriptomics data analysis workflow, called Stereo-seq Analysis Workflow (SAW), was developed for the Stereo-seq technology developed at BGI. SAW includes mRNA spatial position reconstruction, genome alignment, gene expression matrix generation, and clustering. The workflow outputs files in a universal format for subsequent personalized analysis. The execution time for the entire analysis is ∼148 min with 1 GB reads 1 × 1 cm chip test data, 1.8 times faster than with an unoptimized workflow.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
