{"title":"从周期局部耦合簇理论看氧化镁表面 CO 的吸附和振动光谱学","authors":"Hong-Zhou Ye and Timothy C. Berkelbach","doi":"10.1039/D4FD00041B","DOIUrl":null,"url":null,"abstract":"<p >The adsorption of CO on the surface of MgO has long been a model problem in surface chemistry. Here, we report periodic Gaussian-based calculations for this problem using second-order perturbation theory (MP2) and coupled-cluster theory with single and double excitations (CCSD) and perturbative triple excitations [CCSD(T)], with the latter two performed using a recently developed extension of the local natural orbital approximation to problems with periodic boundary conditions. The low cost of periodic local correlation calculations allows us to calculate the full CCSD(T) binding curve of CO approaching the surface of MgO (and thus the adsorption energy) and the two-dimensional potential energy surface (PES) as a function of the distance from the surface and the CO stretching coordinate. From the PES, we obtain the fundamental vibrational frequency of CO on MgO, whose shift from the gas phase value is a common experimental probe of surface adsorption. We find that CCSD(T) correctly predicts a positive frequency shift upon adsorption of +14.7 cm<small><sup>−1</sup></small>, in excellent agreement with the experimental shift of +14.3 cm<small><sup>−1</sup></small>. We use our CCSD(T) results to assess the accuracy of MP2, CCSD, and several density functional theory (DFT) approximations, including exchange correlation functionals and dispersion corrections. We find that MP2 and CCSD yield reasonable binding energies and frequency shifts, whereas many DFT calculations overestimate the magnitude of the adsorption energy by 5–15 kJ mol<small><sup>−1</sup></small> and predict a negative frequency shift of about −20 cm<small><sup>−1</sup></small>, which we attribute to self-interaction-induced delocalization errors that are mildly ameliorated with hybrid functionals. Our findings highlight the accuracy and computational efficiency of the periodic local correlation for the simulation of surface chemistry with accurate wavefunction methods.</p>","PeriodicalId":49075,"journal":{"name":"Faraday Discussions","volume":"254 ","pages":" 628-640"},"PeriodicalIF":3.1000,"publicationDate":"2024-03-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/fd/d4fd00041b?page=search","citationCount":"0","resultStr":"{\"title\":\"Adsorption and vibrational spectroscopy of CO on the surface of MgO from periodic local coupled-cluster theory†\",\"authors\":\"Hong-Zhou Ye and Timothy C. Berkelbach\",\"doi\":\"10.1039/D4FD00041B\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The adsorption of CO on the surface of MgO has long been a model problem in surface chemistry. Here, we report periodic Gaussian-based calculations for this problem using second-order perturbation theory (MP2) and coupled-cluster theory with single and double excitations (CCSD) and perturbative triple excitations [CCSD(T)], with the latter two performed using a recently developed extension of the local natural orbital approximation to problems with periodic boundary conditions. The low cost of periodic local correlation calculations allows us to calculate the full CCSD(T) binding curve of CO approaching the surface of MgO (and thus the adsorption energy) and the two-dimensional potential energy surface (PES) as a function of the distance from the surface and the CO stretching coordinate. From the PES, we obtain the fundamental vibrational frequency of CO on MgO, whose shift from the gas phase value is a common experimental probe of surface adsorption. We find that CCSD(T) correctly predicts a positive frequency shift upon adsorption of +14.7 cm<small><sup>−1</sup></small>, in excellent agreement with the experimental shift of +14.3 cm<small><sup>−1</sup></small>. We use our CCSD(T) results to assess the accuracy of MP2, CCSD, and several density functional theory (DFT) approximations, including exchange correlation functionals and dispersion corrections. We find that MP2 and CCSD yield reasonable binding energies and frequency shifts, whereas many DFT calculations overestimate the magnitude of the adsorption energy by 5–15 kJ mol<small><sup>−1</sup></small> and predict a negative frequency shift of about −20 cm<small><sup>−1</sup></small>, which we attribute to self-interaction-induced delocalization errors that are mildly ameliorated with hybrid functionals. Our findings highlight the accuracy and computational efficiency of the periodic local correlation for the simulation of surface chemistry with accurate wavefunction methods.</p>\",\"PeriodicalId\":49075,\"journal\":{\"name\":\"Faraday Discussions\",\"volume\":\"254 \",\"pages\":\" 628-640\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-03-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/fd/d4fd00041b?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Faraday Discussions\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d4fd00041b\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Faraday Discussions","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d4fd00041b","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Chemistry","Score":null,"Total":0}
Adsorption and vibrational spectroscopy of CO on the surface of MgO from periodic local coupled-cluster theory†
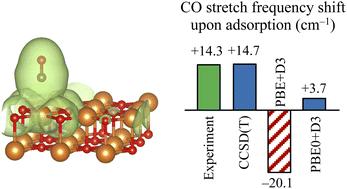
The adsorption of CO on the surface of MgO has long been a model problem in surface chemistry. Here, we report periodic Gaussian-based calculations for this problem using second-order perturbation theory (MP2) and coupled-cluster theory with single and double excitations (CCSD) and perturbative triple excitations [CCSD(T)], with the latter two performed using a recently developed extension of the local natural orbital approximation to problems with periodic boundary conditions. The low cost of periodic local correlation calculations allows us to calculate the full CCSD(T) binding curve of CO approaching the surface of MgO (and thus the adsorption energy) and the two-dimensional potential energy surface (PES) as a function of the distance from the surface and the CO stretching coordinate. From the PES, we obtain the fundamental vibrational frequency of CO on MgO, whose shift from the gas phase value is a common experimental probe of surface adsorption. We find that CCSD(T) correctly predicts a positive frequency shift upon adsorption of +14.7 cm−1, in excellent agreement with the experimental shift of +14.3 cm−1. We use our CCSD(T) results to assess the accuracy of MP2, CCSD, and several density functional theory (DFT) approximations, including exchange correlation functionals and dispersion corrections. We find that MP2 and CCSD yield reasonable binding energies and frequency shifts, whereas many DFT calculations overestimate the magnitude of the adsorption energy by 5–15 kJ mol−1 and predict a negative frequency shift of about −20 cm−1, which we attribute to self-interaction-induced delocalization errors that are mildly ameliorated with hybrid functionals. Our findings highlight the accuracy and computational efficiency of the periodic local correlation for the simulation of surface chemistry with accurate wavefunction methods.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
