Hugo López-Fernández, Miguel Pinto, Cristina P Vieira, Pedro Duque, Miguel Reboiro-Jato, Jorge Vieira
{"title":"Auto-phylo v2 和 aut-phylo-pipeliner:构建先进、灵活和可重复使用的管道,用于系统发育推断、变异性水平估计和正选氨基酸位点识别。","authors":"Hugo López-Fernández, Miguel Pinto, Cristina P Vieira, Pedro Duque, Miguel Reboiro-Jato, Jorge Vieira","doi":"10.1515/jib-2023-0046","DOIUrl":null,"url":null,"abstract":"<p><p>The vast amount of genome sequence data that is available, and that is predicted to drastically increase in the near future, can only be efficiently dealt with by building automated pipelines. Indeed, the Earth Biogenome Project will produce high-quality reference genome sequences for all 1.8 million named living eukaryote species, providing unprecedented insight into the evolution of genes and gene families, and thus on biological issues. Here, new modules for gene annotation, further BLAST search algorithms, further multiple sequence alignment methods, the adding of reference sequences, further tree rooting methods, the estimation of rates of synonymous and nonsynonymous substitutions, and the identification of positively selected amino acid sites, have been added to auto-phylo (version 2), a recently developed software to address biological problems using phylogenetic inferences. Additionally, we present auto-phylo-pipeliner, a graphical user interface application that further facilitates the creation and running of auto-phylo pipelines. Inferences on <i>S-RNase</i> specificity, are critical for both cross-based breeding and for the establishment of pollination requirements. Therefore, as a test case, we develop an auto-phylo pipeline to identify amino acid sites under positive selection, that are, in principle, those determining <i>S-RNase</i> specificity, starting from both non-annotated <i>Prunus</i> genomes and sequences available in public databases.</p>","PeriodicalId":53625,"journal":{"name":"Journal of Integrative Bioinformatics","volume":" ","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2024-03-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11378518/pdf/","citationCount":"0","resultStr":"{\"title\":\"Auto-phylo v2 and auto-phylo-pipeliner: building advanced, flexible, and reusable pipelines for phylogenetic inferences, estimation of variability levels and identification of positively selected amino acid sites.\",\"authors\":\"Hugo López-Fernández, Miguel Pinto, Cristina P Vieira, Pedro Duque, Miguel Reboiro-Jato, Jorge Vieira\",\"doi\":\"10.1515/jib-2023-0046\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The vast amount of genome sequence data that is available, and that is predicted to drastically increase in the near future, can only be efficiently dealt with by building automated pipelines. Indeed, the Earth Biogenome Project will produce high-quality reference genome sequences for all 1.8 million named living eukaryote species, providing unprecedented insight into the evolution of genes and gene families, and thus on biological issues. Here, new modules for gene annotation, further BLAST search algorithms, further multiple sequence alignment methods, the adding of reference sequences, further tree rooting methods, the estimation of rates of synonymous and nonsynonymous substitutions, and the identification of positively selected amino acid sites, have been added to auto-phylo (version 2), a recently developed software to address biological problems using phylogenetic inferences. Additionally, we present auto-phylo-pipeliner, a graphical user interface application that further facilitates the creation and running of auto-phylo pipelines. Inferences on <i>S-RNase</i> specificity, are critical for both cross-based breeding and for the establishment of pollination requirements. Therefore, as a test case, we develop an auto-phylo pipeline to identify amino acid sites under positive selection, that are, in principle, those determining <i>S-RNase</i> specificity, starting from both non-annotated <i>Prunus</i> genomes and sequences available in public databases.</p>\",\"PeriodicalId\":53625,\"journal\":{\"name\":\"Journal of Integrative Bioinformatics\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-03-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11378518/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Integrative Bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1515/jib-2023-0046\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/6/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Integrative Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1515/jib-2023-0046","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Auto-phylo v2 and auto-phylo-pipeliner: building advanced, flexible, and reusable pipelines for phylogenetic inferences, estimation of variability levels and identification of positively selected amino acid sites.
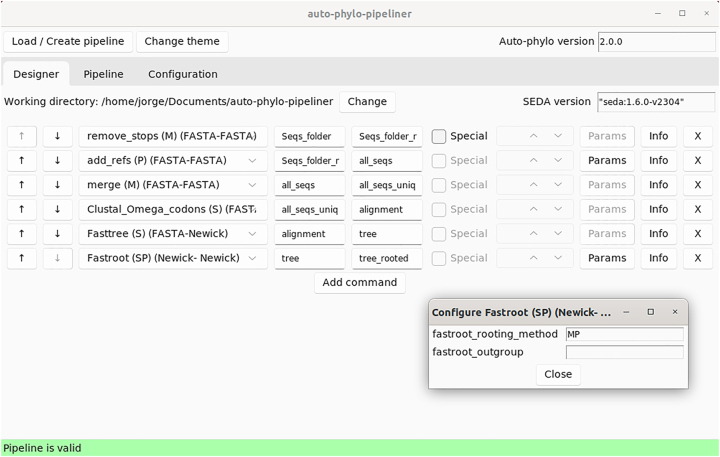
The vast amount of genome sequence data that is available, and that is predicted to drastically increase in the near future, can only be efficiently dealt with by building automated pipelines. Indeed, the Earth Biogenome Project will produce high-quality reference genome sequences for all 1.8 million named living eukaryote species, providing unprecedented insight into the evolution of genes and gene families, and thus on biological issues. Here, new modules for gene annotation, further BLAST search algorithms, further multiple sequence alignment methods, the adding of reference sequences, further tree rooting methods, the estimation of rates of synonymous and nonsynonymous substitutions, and the identification of positively selected amino acid sites, have been added to auto-phylo (version 2), a recently developed software to address biological problems using phylogenetic inferences. Additionally, we present auto-phylo-pipeliner, a graphical user interface application that further facilitates the creation and running of auto-phylo pipelines. Inferences on S-RNase specificity, are critical for both cross-based breeding and for the establishment of pollination requirements. Therefore, as a test case, we develop an auto-phylo pipeline to identify amino acid sites under positive selection, that are, in principle, those determining S-RNase specificity, starting from both non-annotated Prunus genomes and sequences available in public databases.






 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
