Noor Redha, Zahra Al-Sahlawi, Hasan Hasan, Sara Ghareeb, Hani Humaidan
{"title":"线粒体神经胃肠道脑肌病(MNGIE)新型突变引起的脑膜脑炎结束了家族诊断的奥德赛:病例系列报告。","authors":"Noor Redha, Zahra Al-Sahlawi, Hasan Hasan, Sara Ghareeb, Hani Humaidan","doi":"10.1177/11795735241241423","DOIUrl":null,"url":null,"abstract":"<p><p>MNGIE (Mitochondrial Neurogastrointestinal Encephalomyopathy) is an ultra-rare autosomal recessive disorder that leads to mutations in the nuclear genes encoding thymidine phosphorylase. Symptoms include gastrointestinal dysmotility, cachexia, ptosis, external ophthalmoplegia, sensorimotor neuropathy and asymptomatic leukoencephalopathy. We describe the first case of MNGIE with meningoencephalitis that ultimately led to a familial diagnosis ending a diagnostic odyssey. We retrospectively reviewed the electronic medical records and sent whole exome sequencing for the index case and his family members. We report the variant c.877T>C p.(Cys293Arg) found in <i>TYMP</i> gene in all affected siblings showed typical clinical manifestations related to MNGIE. To the best of our knowledge, this is not described in the literature nor in the population databases dbSNP (Single Nucleotide Polymorphism Database) and gnomAD (Genome Aggregation Database). Additionally, it is located in a highly conserved residue and the bioinformatic analysis suggests it is most probably deleterious. Moreover, we estimated 550 number of cases of MNGIE (including 5 cases in this study) after performing an extensive search in the literature across 3 databases from 1983-2023. In addition, we identified 44 patients with MNGIE-like phenotype in genes other than <i>TYMP</i>. MNGIE-like phenotype affects <i>POLG1</i>, <i>RRM2B, LIG3, RRM1, MTTV1</i>, and <i>MT-RNR1</i> genes.</p>","PeriodicalId":15218,"journal":{"name":"Journal of Central Nervous System Disease","volume":"16 ","pages":"11795735241241423"},"PeriodicalIF":2.8000,"publicationDate":"2024-03-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10976485/pdf/","citationCount":"0","resultStr":"{\"title\":\"Meningoencephalitis in a novel mutation in MNGIE (mitochondrial neurogastrointestinal encephalomyopathy) ending a familial diagnostic odyssey: A case series report.\",\"authors\":\"Noor Redha, Zahra Al-Sahlawi, Hasan Hasan, Sara Ghareeb, Hani Humaidan\",\"doi\":\"10.1177/11795735241241423\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>MNGIE (Mitochondrial Neurogastrointestinal Encephalomyopathy) is an ultra-rare autosomal recessive disorder that leads to mutations in the nuclear genes encoding thymidine phosphorylase. Symptoms include gastrointestinal dysmotility, cachexia, ptosis, external ophthalmoplegia, sensorimotor neuropathy and asymptomatic leukoencephalopathy. We describe the first case of MNGIE with meningoencephalitis that ultimately led to a familial diagnosis ending a diagnostic odyssey. We retrospectively reviewed the electronic medical records and sent whole exome sequencing for the index case and his family members. We report the variant c.877T>C p.(Cys293Arg) found in <i>TYMP</i> gene in all affected siblings showed typical clinical manifestations related to MNGIE. To the best of our knowledge, this is not described in the literature nor in the population databases dbSNP (Single Nucleotide Polymorphism Database) and gnomAD (Genome Aggregation Database). Additionally, it is located in a highly conserved residue and the bioinformatic analysis suggests it is most probably deleterious. Moreover, we estimated 550 number of cases of MNGIE (including 5 cases in this study) after performing an extensive search in the literature across 3 databases from 1983-2023. In addition, we identified 44 patients with MNGIE-like phenotype in genes other than <i>TYMP</i>. MNGIE-like phenotype affects <i>POLG1</i>, <i>RRM2B, LIG3, RRM1, MTTV1</i>, and <i>MT-RNR1</i> genes.</p>\",\"PeriodicalId\":15218,\"journal\":{\"name\":\"Journal of Central Nervous System Disease\",\"volume\":\"16 \",\"pages\":\"11795735241241423\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-03-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10976485/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Central Nervous System Disease\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11795735241241423\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Central Nervous System Disease","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11795735241241423","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Meningoencephalitis in a novel mutation in MNGIE (mitochondrial neurogastrointestinal encephalomyopathy) ending a familial diagnostic odyssey: A case series report.
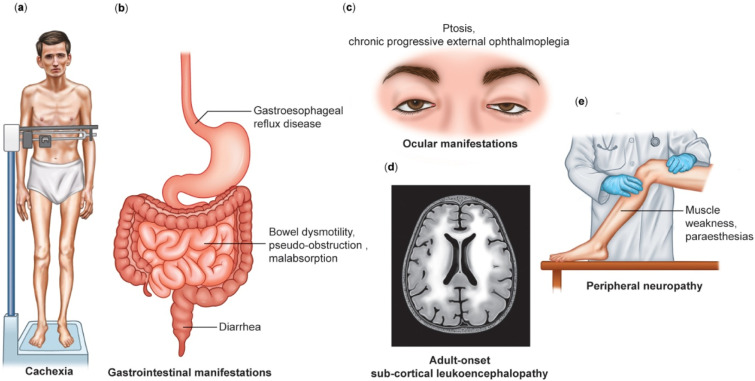
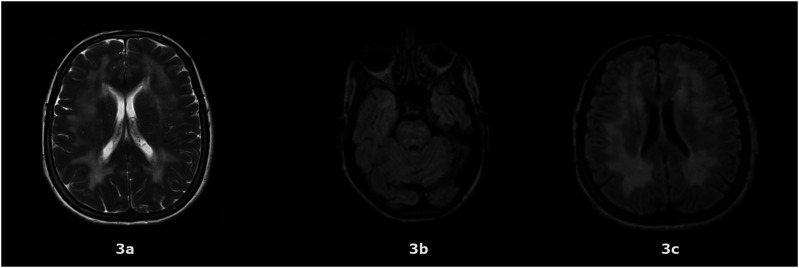
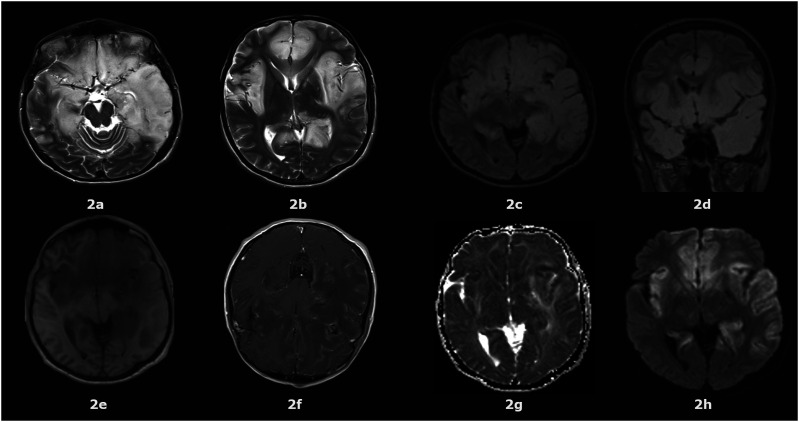
MNGIE (Mitochondrial Neurogastrointestinal Encephalomyopathy) is an ultra-rare autosomal recessive disorder that leads to mutations in the nuclear genes encoding thymidine phosphorylase. Symptoms include gastrointestinal dysmotility, cachexia, ptosis, external ophthalmoplegia, sensorimotor neuropathy and asymptomatic leukoencephalopathy. We describe the first case of MNGIE with meningoencephalitis that ultimately led to a familial diagnosis ending a diagnostic odyssey. We retrospectively reviewed the electronic medical records and sent whole exome sequencing for the index case and his family members. We report the variant c.877T>C p.(Cys293Arg) found in TYMP gene in all affected siblings showed typical clinical manifestations related to MNGIE. To the best of our knowledge, this is not described in the literature nor in the population databases dbSNP (Single Nucleotide Polymorphism Database) and gnomAD (Genome Aggregation Database). Additionally, it is located in a highly conserved residue and the bioinformatic analysis suggests it is most probably deleterious. Moreover, we estimated 550 number of cases of MNGIE (including 5 cases in this study) after performing an extensive search in the literature across 3 databases from 1983-2023. In addition, we identified 44 patients with MNGIE-like phenotype in genes other than TYMP. MNGIE-like phenotype affects POLG1, RRM2B, LIG3, RRM1, MTTV1, and MT-RNR1 genes.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
