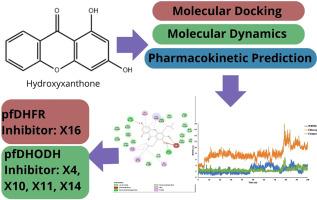
{"title":"通过分子对接、分子动力学模拟、MM-PBSA 计算和药代动力学预测,对羟基氧杂蒽酮衍生物作为潜在 pfDHFR 和 pfDHODH 抑制剂的分子内研究","authors":"Lathifah Puji Hastuti , Faris Hermawan , Muthia Rahayu Iresha , Teni Ernawati , Firdayani","doi":"10.1016/j.imu.2024.101485","DOIUrl":null,"url":null,"abstract":"<div><p>The investigation of hydroxyxanthone derivatives has been conducted, including molecular docking, molecular dynamics simulation MM-PBSA binding energy calculation, and pharmacokinetics prediction of the potential <em>plasmodium falciparum</em> dihydrofolate reductase (<em>pf</em>DHFR) and <em>plasmodium falciparum</em> dihydroorotate dehydrogenase (<em>pf</em>DHODH) inhibitor. The Docking result showed that compound 1,3,6,7-tetrahydroxy-5,8-bis(3-methyl-2-buten-1-yl)-9H-xanthen-9-one (<strong>X16</strong>) was found to be the best ligand with good inhibitory action against <em>pf</em>DHFR. Meanwhile, the <em>pf</em>DHODH protein was compounded 1,3,6,7-tetrahydroxy-5,8-dinitro-9H-xanthen-9-one (<strong>X14</strong>). Additionally, the hydroxyxanthone <strong>X16</strong> complex showed more excellent stability in the molecular dynamics simulation of the <em>pf</em>DHFR protein than the ligand WR99210 and chloroquine. The MM-PBSA calculation showed that compound <strong>X16</strong> had lower binding energy than ligand WR99210. However, 1,3-dihydroxy-8-(3-methyl-2-buten-1-yl)-9H-xanthen-9-one <strong>(X4)</strong>, 1,3,6,7-tetrahydroxy-8-nitro-9H-xanthen-9-one (<strong>X10),</strong> 1,3,6,7-tetrahydroxy-9-oxo-9H-xanthene-8-sulfonic acid <strong>(X11)</strong>, and 1,3,6,7-tetrahydroxy-5,8-dinitro-9H-xanthen-9-one <strong>(X14)</strong> complexes were shown to be more stable than chloroquine and to have the same stability when compared to the native ligand A26, according to a molecular dynamics simulation conducted in <em>pf</em>DHODH protein. The MM-PBSA calculation showed that compound <strong>X14</strong> had lower binding energy than ligand A26. The hydroxyxanthones <strong>X4</strong>, <strong>X10</strong>–<strong>11</strong>, <strong>X14</strong>, and <strong>X16</strong> fulfill Lipinski's rule parameters in terms of physicochemical and ADMET qualities and parameters related to absorption, distribution, metabolism, excretion, and toxicity tests. To sum up, hydroxyxanthones <strong>X4</strong>, <strong>X10</strong>–<strong>11</strong>, <strong>X14</strong>, and <strong>X16</strong> have the potential to be antimalarial medications, but more in vivo and in vitro testing is needed to confirm this.</p></div>","PeriodicalId":13953,"journal":{"name":"Informatics in Medicine Unlocked","volume":"47 ","pages":"Article 101485"},"PeriodicalIF":0.0000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2352914824000418/pdfft?md5=f5f76f623d6bb9357e5af1d58934327c&pid=1-s2.0-S2352914824000418-main.pdf","citationCount":"0","resultStr":"{\"title\":\"In-silico studies of hydroxyxanthone derivatives as potential pfDHFR and pfDHODH inhibitor by molecular docking, molecular dynamics simulation, MM-PBSA calculation and pharmacokinetics prediction\",\"authors\":\"Lathifah Puji Hastuti , Faris Hermawan , Muthia Rahayu Iresha , Teni Ernawati , Firdayani\",\"doi\":\"10.1016/j.imu.2024.101485\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The investigation of hydroxyxanthone derivatives has been conducted, including molecular docking, molecular dynamics simulation MM-PBSA binding energy calculation, and pharmacokinetics prediction of the potential <em>plasmodium falciparum</em> dihydrofolate reductase (<em>pf</em>DHFR) and <em>plasmodium falciparum</em> dihydroorotate dehydrogenase (<em>pf</em>DHODH) inhibitor. The Docking result showed that compound 1,3,6,7-tetrahydroxy-5,8-bis(3-methyl-2-buten-1-yl)-9H-xanthen-9-one (<strong>X16</strong>) was found to be the best ligand with good inhibitory action against <em>pf</em>DHFR. Meanwhile, the <em>pf</em>DHODH protein was compounded 1,3,6,7-tetrahydroxy-5,8-dinitro-9H-xanthen-9-one (<strong>X14</strong>). Additionally, the hydroxyxanthone <strong>X16</strong> complex showed more excellent stability in the molecular dynamics simulation of the <em>pf</em>DHFR protein than the ligand WR99210 and chloroquine. The MM-PBSA calculation showed that compound <strong>X16</strong> had lower binding energy than ligand WR99210. However, 1,3-dihydroxy-8-(3-methyl-2-buten-1-yl)-9H-xanthen-9-one <strong>(X4)</strong>, 1,3,6,7-tetrahydroxy-8-nitro-9H-xanthen-9-one (<strong>X10),</strong> 1,3,6,7-tetrahydroxy-9-oxo-9H-xanthene-8-sulfonic acid <strong>(X11)</strong>, and 1,3,6,7-tetrahydroxy-5,8-dinitro-9H-xanthen-9-one <strong>(X14)</strong> complexes were shown to be more stable than chloroquine and to have the same stability when compared to the native ligand A26, according to a molecular dynamics simulation conducted in <em>pf</em>DHODH protein. The MM-PBSA calculation showed that compound <strong>X14</strong> had lower binding energy than ligand A26. The hydroxyxanthones <strong>X4</strong>, <strong>X10</strong>–<strong>11</strong>, <strong>X14</strong>, and <strong>X16</strong> fulfill Lipinski's rule parameters in terms of physicochemical and ADMET qualities and parameters related to absorption, distribution, metabolism, excretion, and toxicity tests. To sum up, hydroxyxanthones <strong>X4</strong>, <strong>X10</strong>–<strong>11</strong>, <strong>X14</strong>, and <strong>X16</strong> have the potential to be antimalarial medications, but more in vivo and in vitro testing is needed to confirm this.</p></div>\",\"PeriodicalId\":13953,\"journal\":{\"name\":\"Informatics in Medicine Unlocked\",\"volume\":\"47 \",\"pages\":\"Article 101485\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2352914824000418/pdfft?md5=f5f76f623d6bb9357e5af1d58934327c&pid=1-s2.0-S2352914824000418-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Informatics in Medicine Unlocked\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2352914824000418\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/3/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Informatics in Medicine Unlocked","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2352914824000418","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/27 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
In-silico studies of hydroxyxanthone derivatives as potential pfDHFR and pfDHODH inhibitor by molecular docking, molecular dynamics simulation, MM-PBSA calculation and pharmacokinetics prediction
The investigation of hydroxyxanthone derivatives has been conducted, including molecular docking, molecular dynamics simulation MM-PBSA binding energy calculation, and pharmacokinetics prediction of the potential plasmodium falciparum dihydrofolate reductase (pfDHFR) and plasmodium falciparum dihydroorotate dehydrogenase (pfDHODH) inhibitor. The Docking result showed that compound 1,3,6,7-tetrahydroxy-5,8-bis(3-methyl-2-buten-1-yl)-9H-xanthen-9-one (X16) was found to be the best ligand with good inhibitory action against pfDHFR. Meanwhile, the pfDHODH protein was compounded 1,3,6,7-tetrahydroxy-5,8-dinitro-9H-xanthen-9-one (X14). Additionally, the hydroxyxanthone X16 complex showed more excellent stability in the molecular dynamics simulation of the pfDHFR protein than the ligand WR99210 and chloroquine. The MM-PBSA calculation showed that compound X16 had lower binding energy than ligand WR99210. However, 1,3-dihydroxy-8-(3-methyl-2-buten-1-yl)-9H-xanthen-9-one (X4), 1,3,6,7-tetrahydroxy-8-nitro-9H-xanthen-9-one (X10), 1,3,6,7-tetrahydroxy-9-oxo-9H-xanthene-8-sulfonic acid (X11), and 1,3,6,7-tetrahydroxy-5,8-dinitro-9H-xanthen-9-one (X14) complexes were shown to be more stable than chloroquine and to have the same stability when compared to the native ligand A26, according to a molecular dynamics simulation conducted in pfDHODH protein. The MM-PBSA calculation showed that compound X14 had lower binding energy than ligand A26. The hydroxyxanthones X4, X10–11, X14, and X16 fulfill Lipinski's rule parameters in terms of physicochemical and ADMET qualities and parameters related to absorption, distribution, metabolism, excretion, and toxicity tests. To sum up, hydroxyxanthones X4, X10–11, X14, and X16 have the potential to be antimalarial medications, but more in vivo and in vitro testing is needed to confirm this.
期刊介绍:
Informatics in Medicine Unlocked (IMU) is an international gold open access journal covering a broad spectrum of topics within medical informatics, including (but not limited to) papers focusing on imaging, pathology, teledermatology, public health, ophthalmological, nursing and translational medicine informatics. The full papers that are published in the journal are accessible to all who visit the website.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
