{"title":"通过HMGB1 mRNA降解介导的STAT3/DUSP16轴调控,平滑肌细胞ILF3的缺乏可减轻内膜增生","authors":"Ya-min Hou, Bo-han Xu, Qiu-ting Zhang, Jie Cheng, Xu Zhang, Hong-rui Yang, Ze-ying Wang, Peng Wang, Ming-xiang Zhang","doi":"10.1016/j.yjmcc.2024.04.004","DOIUrl":null,"url":null,"abstract":"<div><p>Intimal hyperplasia is a complicated pathophysiological phenomenon attributable to in-stent restenosis, and the underlying mechanism remains unclear. Interleukin enhancer-binding factor 3 (ILF3), a double-stranded RNA-binding protein involved in regulating mRNA stability, has been recently demonstrated to assume a crucial role in cardiovascular disease; nevertheless, its impact on intimal hyperplasia remains unknown. In current study, we used samples of human restenotic arteries and rodent models of intimal hyperplasia, we found that vascular smooth muscle cell (VSMC) ILF3 expression was markedly elevated in human restenotic arteries and murine ligated carotid arteries. SMC-specific ILF3 knockout mice significantly suppressed injury induced neointimal formation. In vitro, platelet-derived growth factor type BB (PDGF-BB) treatment elevated the level of VSMC ILF3 in a dose- and time-dependent manner. ILF3 silencing markedly inhibited PDGF-BB-induced phenotype switching, proliferation, and migration in VSMCs. Transcriptome sequencing and RNA immunoprecipitation sequencing depicted that ILF3 maintained its stability upon binding to the mRNA of the high-mobility group box 1 protein (HMGB1), thereby exerting an inhibitory effect on the transcription of dual specificity phosphatase 16 (DUSP16) through enhanced phosphorylation of signal transducer and activator of transcription 3 (STAT3). Therefore, the results both in vitro and in vivo indicated that the loss of ILF3 in VSMC ameliorated neointimal hyperplasia by regulating the STAT3/DUSP16 axis through the degradation of HMGB1 mRNA. Our findings revealed that vascular injury activates VSMC ILF3, which in turn promotes intima formation. Consequently, targeting specific VSMC ILF3 may present a potential therapeutic strategy for ameliorating cardiovascular restenosis.</p></div>","PeriodicalId":16402,"journal":{"name":"Journal of molecular and cellular cardiology","volume":"190 ","pages":"Pages 62-75"},"PeriodicalIF":4.7000,"publicationDate":"2024-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Deficiency of smooth muscle cell ILF3 alleviates intimal hyperplasia via HMGB1 mRNA degradation-mediated regulation of the STAT3/DUSP16 axis\",\"authors\":\"Ya-min Hou, Bo-han Xu, Qiu-ting Zhang, Jie Cheng, Xu Zhang, Hong-rui Yang, Ze-ying Wang, Peng Wang, Ming-xiang Zhang\",\"doi\":\"10.1016/j.yjmcc.2024.04.004\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Intimal hyperplasia is a complicated pathophysiological phenomenon attributable to in-stent restenosis, and the underlying mechanism remains unclear. Interleukin enhancer-binding factor 3 (ILF3), a double-stranded RNA-binding protein involved in regulating mRNA stability, has been recently demonstrated to assume a crucial role in cardiovascular disease; nevertheless, its impact on intimal hyperplasia remains unknown. In current study, we used samples of human restenotic arteries and rodent models of intimal hyperplasia, we found that vascular smooth muscle cell (VSMC) ILF3 expression was markedly elevated in human restenotic arteries and murine ligated carotid arteries. SMC-specific ILF3 knockout mice significantly suppressed injury induced neointimal formation. In vitro, platelet-derived growth factor type BB (PDGF-BB) treatment elevated the level of VSMC ILF3 in a dose- and time-dependent manner. ILF3 silencing markedly inhibited PDGF-BB-induced phenotype switching, proliferation, and migration in VSMCs. Transcriptome sequencing and RNA immunoprecipitation sequencing depicted that ILF3 maintained its stability upon binding to the mRNA of the high-mobility group box 1 protein (HMGB1), thereby exerting an inhibitory effect on the transcription of dual specificity phosphatase 16 (DUSP16) through enhanced phosphorylation of signal transducer and activator of transcription 3 (STAT3). Therefore, the results both in vitro and in vivo indicated that the loss of ILF3 in VSMC ameliorated neointimal hyperplasia by regulating the STAT3/DUSP16 axis through the degradation of HMGB1 mRNA. Our findings revealed that vascular injury activates VSMC ILF3, which in turn promotes intima formation. Consequently, targeting specific VSMC ILF3 may present a potential therapeutic strategy for ameliorating cardiovascular restenosis.</p></div>\",\"PeriodicalId\":16402,\"journal\":{\"name\":\"Journal of molecular and cellular cardiology\",\"volume\":\"190 \",\"pages\":\"Pages 62-75\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2024-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular and cellular cardiology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0022282824000488\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/4/6 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CARDIAC & CARDIOVASCULAR SYSTEMS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular and cellular cardiology","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022282824000488","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/4/6 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CARDIAC & CARDIOVASCULAR SYSTEMS","Score":null,"Total":0}
Deficiency of smooth muscle cell ILF3 alleviates intimal hyperplasia via HMGB1 mRNA degradation-mediated regulation of the STAT3/DUSP16 axis
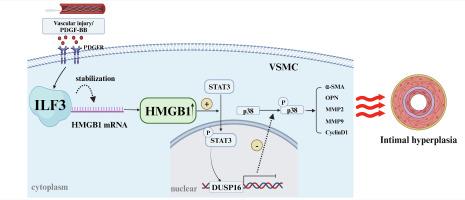
Intimal hyperplasia is a complicated pathophysiological phenomenon attributable to in-stent restenosis, and the underlying mechanism remains unclear. Interleukin enhancer-binding factor 3 (ILF3), a double-stranded RNA-binding protein involved in regulating mRNA stability, has been recently demonstrated to assume a crucial role in cardiovascular disease; nevertheless, its impact on intimal hyperplasia remains unknown. In current study, we used samples of human restenotic arteries and rodent models of intimal hyperplasia, we found that vascular smooth muscle cell (VSMC) ILF3 expression was markedly elevated in human restenotic arteries and murine ligated carotid arteries. SMC-specific ILF3 knockout mice significantly suppressed injury induced neointimal formation. In vitro, platelet-derived growth factor type BB (PDGF-BB) treatment elevated the level of VSMC ILF3 in a dose- and time-dependent manner. ILF3 silencing markedly inhibited PDGF-BB-induced phenotype switching, proliferation, and migration in VSMCs. Transcriptome sequencing and RNA immunoprecipitation sequencing depicted that ILF3 maintained its stability upon binding to the mRNA of the high-mobility group box 1 protein (HMGB1), thereby exerting an inhibitory effect on the transcription of dual specificity phosphatase 16 (DUSP16) through enhanced phosphorylation of signal transducer and activator of transcription 3 (STAT3). Therefore, the results both in vitro and in vivo indicated that the loss of ILF3 in VSMC ameliorated neointimal hyperplasia by regulating the STAT3/DUSP16 axis through the degradation of HMGB1 mRNA. Our findings revealed that vascular injury activates VSMC ILF3, which in turn promotes intima formation. Consequently, targeting specific VSMC ILF3 may present a potential therapeutic strategy for ameliorating cardiovascular restenosis.
期刊介绍:
The Journal of Molecular and Cellular Cardiology publishes work advancing knowledge of the mechanisms responsible for both normal and diseased cardiovascular function. To this end papers are published in all relevant areas. These include (but are not limited to): structural biology; genetics; proteomics; morphology; stem cells; molecular biology; metabolism; biophysics; bioengineering; computational modeling and systems analysis; electrophysiology; pharmacology and physiology. Papers are encouraged with both basic and translational approaches. The journal is directed not only to basic scientists but also to clinical cardiologists who wish to follow the rapidly advancing frontiers of basic knowledge of the heart and circulation.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
