Alexandra Klein, Katharina Klug, Maximilian Breyer, Julia Grüner, Vijay Krishna Medala, Peter Nordbeck, Christoph Wanner, Eva Klopocki, Nurcan Üçeyler
{"title":"意义不明的α-半乳糖苷酶 A 基因变异:法布里病的细胞划分","authors":"Alexandra Klein, Katharina Klug, Maximilian Breyer, Julia Grüner, Vijay Krishna Medala, Peter Nordbeck, Christoph Wanner, Eva Klopocki, Nurcan Üçeyler","doi":"10.1002/jimd.12743","DOIUrl":null,"url":null,"abstract":"<p>Fabry disease (FD) is an X-linked multiorgan disorder caused by variants in the alpha-galactosidase A gene (<i>GLA</i>). Depending on the variant, disease phenotypes range from benign to life-threatening. More than 1000 <i>GLA</i> variants are known, but a link between genotype and phenotype in FD has not yet been established for all. p.A143T, p.D313Y, and p.S126G are frequent examples of variants of unknown significance (VUS). We have investigated the potential pathogenicity of these VUS combining clinical data with data obtained in human cellular in vitro systems. We have analyzed four different male subject-derived cell types for alpha-galactosidase A enzyme (GLA) activity and intracellular Gb3 load. Additionally, Gb3 load in skin tissue as well as clinical data were studied for correlates of disease manifestations. A reduction of GLA activity was observed in cells carrying p.A143T compared with controls (<i>p</i> < 0.05). In cells carrying the p.D313Y variant, a reduced GLA activity was found only in endothelial cells (<i>p</i> < 0.01) compared with controls. No pathological changes were observed in cells carrying the p.S126G variant. None of the VUS investigated caused intracellular Gb3 accumulation in any cell type. Our data of aberrant GLA activity in cells of p.A143T hemizygotes and overall normal cellular phenotypes in cells of p.D313Y and p.S126G hemizygotes contribute a basic science perspective to the clinically highly relevant discussion on VUS in <i>GLA</i>.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 4","pages":"805-817"},"PeriodicalIF":3.8000,"publicationDate":"2024-04-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12743","citationCount":"0","resultStr":"{\"title\":\"Genetic variants of unknown significance in alpha-galactosidase A: Cellular delineation from Fabry disease\",\"authors\":\"Alexandra Klein, Katharina Klug, Maximilian Breyer, Julia Grüner, Vijay Krishna Medala, Peter Nordbeck, Christoph Wanner, Eva Klopocki, Nurcan Üçeyler\",\"doi\":\"10.1002/jimd.12743\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Fabry disease (FD) is an X-linked multiorgan disorder caused by variants in the alpha-galactosidase A gene (<i>GLA</i>). Depending on the variant, disease phenotypes range from benign to life-threatening. More than 1000 <i>GLA</i> variants are known, but a link between genotype and phenotype in FD has not yet been established for all. p.A143T, p.D313Y, and p.S126G are frequent examples of variants of unknown significance (VUS). We have investigated the potential pathogenicity of these VUS combining clinical data with data obtained in human cellular in vitro systems. We have analyzed four different male subject-derived cell types for alpha-galactosidase A enzyme (GLA) activity and intracellular Gb3 load. Additionally, Gb3 load in skin tissue as well as clinical data were studied for correlates of disease manifestations. A reduction of GLA activity was observed in cells carrying p.A143T compared with controls (<i>p</i> < 0.05). In cells carrying the p.D313Y variant, a reduced GLA activity was found only in endothelial cells (<i>p</i> < 0.01) compared with controls. No pathological changes were observed in cells carrying the p.S126G variant. None of the VUS investigated caused intracellular Gb3 accumulation in any cell type. Our data of aberrant GLA activity in cells of p.A143T hemizygotes and overall normal cellular phenotypes in cells of p.D313Y and p.S126G hemizygotes contribute a basic science perspective to the clinically highly relevant discussion on VUS in <i>GLA</i>.</p>\",\"PeriodicalId\":16281,\"journal\":{\"name\":\"Journal of Inherited Metabolic Disease\",\"volume\":\"47 4\",\"pages\":\"805-817\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2024-04-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12743\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Inherited Metabolic Disease\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12743\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12743","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Genetic variants of unknown significance in alpha-galactosidase A: Cellular delineation from Fabry disease
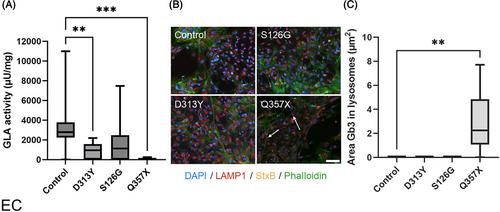
Fabry disease (FD) is an X-linked multiorgan disorder caused by variants in the alpha-galactosidase A gene (GLA). Depending on the variant, disease phenotypes range from benign to life-threatening. More than 1000 GLA variants are known, but a link between genotype and phenotype in FD has not yet been established for all. p.A143T, p.D313Y, and p.S126G are frequent examples of variants of unknown significance (VUS). We have investigated the potential pathogenicity of these VUS combining clinical data with data obtained in human cellular in vitro systems. We have analyzed four different male subject-derived cell types for alpha-galactosidase A enzyme (GLA) activity and intracellular Gb3 load. Additionally, Gb3 load in skin tissue as well as clinical data were studied for correlates of disease manifestations. A reduction of GLA activity was observed in cells carrying p.A143T compared with controls (p < 0.05). In cells carrying the p.D313Y variant, a reduced GLA activity was found only in endothelial cells (p < 0.01) compared with controls. No pathological changes were observed in cells carrying the p.S126G variant. None of the VUS investigated caused intracellular Gb3 accumulation in any cell type. Our data of aberrant GLA activity in cells of p.A143T hemizygotes and overall normal cellular phenotypes in cells of p.D313Y and p.S126G hemizygotes contribute a basic science perspective to the clinically highly relevant discussion on VUS in GLA.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
