{"title":"RAS病的自闭症谱系障碍特征:系统综述","authors":"Edward Debbaut, Jean Steyaert, Mouna El Bakkali","doi":"10.1002/mgg3.2428","DOIUrl":null,"url":null,"abstract":"BackgroundRASopathies are associated with an increased risk of autism spectrum disorder (ASD). For neurofibromatosis type 1 (NF1) there is ample evidence for this increased risk, while for other RASopathies this association has been studied less. No specific ASD profile has been delineated so far for RASopathies or a specific RASopathy individually.MethodsWe conducted a systematic review to investigate whether a specific RASopathy is associated with a specific ASD profile, or if RASopathies altogether have a distinct ASD profile compared to idiopathic ASD (iASD). We searched PubMed, Web of Science, and Open Grey for data about ASD features in RASopathies and potential modifiers.ResultsWe included 41 articles on ASD features in NF1, Noonan syndrome (NS), Costello syndrome (CS), and cardio‐facio‐cutaneous syndrome (CFC). Individuals with NF1, NS, CS, and CFC on average have higher ASD symptomatology than healthy controls and unaffected siblings, though less than people with iASD. There is insufficient evidence for a distinct ASD phenotype in RASopathies compared to iASD or when RASopathies are compared with each other. We identified several potentially modifying factors of ASD symptoms in RASopathies.ConclusionsOur systematic review found no convincing evidence for a specific ASD profile in RASopathies compared to iASD, or in a specific RASopathy compared to other RASopathies. However, we identified important limitations in the research literature which may also account for this result. These limitations are discussed and recommendations for future research are formulated.","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"208 1","pages":""},"PeriodicalIF":1.6000,"publicationDate":"2024-04-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Autism spectrum disorder profiles in RASopathies: A systematic review\",\"authors\":\"Edward Debbaut, Jean Steyaert, Mouna El Bakkali\",\"doi\":\"10.1002/mgg3.2428\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"BackgroundRASopathies are associated with an increased risk of autism spectrum disorder (ASD). For neurofibromatosis type 1 (NF1) there is ample evidence for this increased risk, while for other RASopathies this association has been studied less. No specific ASD profile has been delineated so far for RASopathies or a specific RASopathy individually.MethodsWe conducted a systematic review to investigate whether a specific RASopathy is associated with a specific ASD profile, or if RASopathies altogether have a distinct ASD profile compared to idiopathic ASD (iASD). We searched PubMed, Web of Science, and Open Grey for data about ASD features in RASopathies and potential modifiers.ResultsWe included 41 articles on ASD features in NF1, Noonan syndrome (NS), Costello syndrome (CS), and cardio‐facio‐cutaneous syndrome (CFC). Individuals with NF1, NS, CS, and CFC on average have higher ASD symptomatology than healthy controls and unaffected siblings, though less than people with iASD. There is insufficient evidence for a distinct ASD phenotype in RASopathies compared to iASD or when RASopathies are compared with each other. We identified several potentially modifying factors of ASD symptoms in RASopathies.ConclusionsOur systematic review found no convincing evidence for a specific ASD profile in RASopathies compared to iASD, or in a specific RASopathy compared to other RASopathies. However, we identified important limitations in the research literature which may also account for this result. These limitations are discussed and recommendations for future research are formulated.\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":\"208 1\",\"pages\":\"\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2024-04-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.2428\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.2428","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Autism spectrum disorder profiles in RASopathies: A systematic review
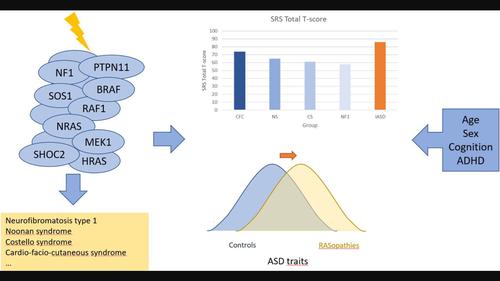
BackgroundRASopathies are associated with an increased risk of autism spectrum disorder (ASD). For neurofibromatosis type 1 (NF1) there is ample evidence for this increased risk, while for other RASopathies this association has been studied less. No specific ASD profile has been delineated so far for RASopathies or a specific RASopathy individually.MethodsWe conducted a systematic review to investigate whether a specific RASopathy is associated with a specific ASD profile, or if RASopathies altogether have a distinct ASD profile compared to idiopathic ASD (iASD). We searched PubMed, Web of Science, and Open Grey for data about ASD features in RASopathies and potential modifiers.ResultsWe included 41 articles on ASD features in NF1, Noonan syndrome (NS), Costello syndrome (CS), and cardio‐facio‐cutaneous syndrome (CFC). Individuals with NF1, NS, CS, and CFC on average have higher ASD symptomatology than healthy controls and unaffected siblings, though less than people with iASD. There is insufficient evidence for a distinct ASD phenotype in RASopathies compared to iASD or when RASopathies are compared with each other. We identified several potentially modifying factors of ASD symptoms in RASopathies.ConclusionsOur systematic review found no convincing evidence for a specific ASD profile in RASopathies compared to iASD, or in a specific RASopathy compared to other RASopathies. However, we identified important limitations in the research literature which may also account for this result. These limitations are discussed and recommendations for future research are formulated.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
