Jun Hee Cho, Soojin Hwang, Yoon Hae Kwak, Mi‐Sun Yum, Go Hun Seo, June‐Young Koh, Young Seok Ju, Ji‐Hee Yoon, Minji Kang, Hyo‐Sang Do, Soyoung Kim, Gu‐Hwan Kim, Hyunwoo Bae, Beom Hee Lee
{"title":"三例先天性痛觉不敏感合并无汗症患者的临床和遗传特征:病例报告和文献综述","authors":"Jun Hee Cho, Soojin Hwang, Yoon Hae Kwak, Mi‐Sun Yum, Go Hun Seo, June‐Young Koh, Young Seok Ju, Ji‐Hee Yoon, Minji Kang, Hyo‐Sang Do, Soyoung Kim, Gu‐Hwan Kim, Hyunwoo Bae, Beom Hee Lee","doi":"10.1002/mgg3.2430","DOIUrl":null,"url":null,"abstract":"BackgroundCongenital insensitivity to pain with anhidrosis (CIPA) is an extremely rare autosomal recessive disorder caused by loss‐of‐function mutations of the <jats:italic>NTRK1</jats:italic> gene, affecting the autonomic and sensory nervous system. Clinical manifestation is varied and includes recurrent fever, pain insensitivity, anhidrosis, self‐mutilating behavior, and intellectual disability.MethodsClinical and genetic features were assessed in two males and one female with genetically confirmed CIPA using exome or genome sequencing.ResultsCIPA symptoms including recurrent fever, pain insensitivity, and anhidrosis manifested at the age of 1 year (age range: 0.3–8 years). Two patients exhibited self‐mutilation tendencies, intellectual disability, and developmental delay. Four <jats:italic>NTRK1</jats:italic> (NM_002529.3) mutations, c.851‐33T>A (p.?), c.2020G>T (p.Asp674Tyr), c.2303C>T (p.Pro768Leu), and c.574‐156_850+1113del (exons 5‐7 del) were identified. Two patients exhibited early onset and severe phenotype, being homozygous for c.851‐33T>A (p.?) mutations and compound heterozygous for c.851‐33T>A (p.?) and c.2020G>T (p.Asp674Tyr) mutation of <jats:italic>NTRK1</jats:italic>. The third patient with compound heterozygous mutations of c.2303C>T (p.Pro768Leu) and c.574‐156_850+1113del (exons 5‐7 del) displayed a late onset and milder clinical manifestation.ConclusionAll three patients exhibited variable phenotypes and disease severity. This research enriches our understanding of clinical and genetic aspects of CIPA, highlighting variable phenotypes and disease severity.","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"59 1","pages":""},"PeriodicalIF":1.6000,"publicationDate":"2024-04-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Clinical and genetic characteristics of three patients with congenital insensitivity to pain with anhidrosis: Case reports and a review of the literature\",\"authors\":\"Jun Hee Cho, Soojin Hwang, Yoon Hae Kwak, Mi‐Sun Yum, Go Hun Seo, June‐Young Koh, Young Seok Ju, Ji‐Hee Yoon, Minji Kang, Hyo‐Sang Do, Soyoung Kim, Gu‐Hwan Kim, Hyunwoo Bae, Beom Hee Lee\",\"doi\":\"10.1002/mgg3.2430\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"BackgroundCongenital insensitivity to pain with anhidrosis (CIPA) is an extremely rare autosomal recessive disorder caused by loss‐of‐function mutations of the <jats:italic>NTRK1</jats:italic> gene, affecting the autonomic and sensory nervous system. Clinical manifestation is varied and includes recurrent fever, pain insensitivity, anhidrosis, self‐mutilating behavior, and intellectual disability.MethodsClinical and genetic features were assessed in two males and one female with genetically confirmed CIPA using exome or genome sequencing.ResultsCIPA symptoms including recurrent fever, pain insensitivity, and anhidrosis manifested at the age of 1 year (age range: 0.3–8 years). Two patients exhibited self‐mutilation tendencies, intellectual disability, and developmental delay. Four <jats:italic>NTRK1</jats:italic> (NM_002529.3) mutations, c.851‐33T>A (p.?), c.2020G>T (p.Asp674Tyr), c.2303C>T (p.Pro768Leu), and c.574‐156_850+1113del (exons 5‐7 del) were identified. Two patients exhibited early onset and severe phenotype, being homozygous for c.851‐33T>A (p.?) mutations and compound heterozygous for c.851‐33T>A (p.?) and c.2020G>T (p.Asp674Tyr) mutation of <jats:italic>NTRK1</jats:italic>. The third patient with compound heterozygous mutations of c.2303C>T (p.Pro768Leu) and c.574‐156_850+1113del (exons 5‐7 del) displayed a late onset and milder clinical manifestation.ConclusionAll three patients exhibited variable phenotypes and disease severity. This research enriches our understanding of clinical and genetic aspects of CIPA, highlighting variable phenotypes and disease severity.\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":\"59 1\",\"pages\":\"\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2024-04-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.2430\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.2430","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
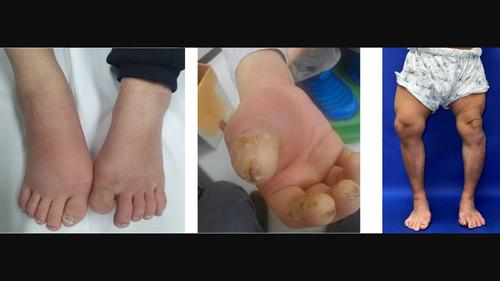
背景先天性痛觉不敏感伴潮湿症(CIPA)是一种极其罕见的常染色体隐性遗传疾病,由NTRK1基因功能缺失突变引起,影响自主神经和感觉神经系统。临床表现多种多样,包括反复发热、对疼痛不敏感、多汗症、自残行为和智力障碍。两名患者有自残倾向、智力障碍和发育迟缓。发现了四种 NTRK1(NM_002529.3)突变,分别为 c.851-33T>A(p.?)、c.2020G>T(p.Asp674Tyr)、c.2303C>T(p.Pro768Leu)和 c.574-156_850+1113del(5-7 del 外显子)。两名患者表现出早发和严重的表型,分别是NTRK1的c.851-33T>A(p.?)突变的同源杂合子和c.851-33T>A(p.?)和c.2020G>T(p.Asp674Tyr)突变的复合杂合子。第三位患者为c.2303C>T(p.Pro768Leu)和c.574-156_850+1113del(5-7 del外显子)复合杂合突变,起病较晚,临床表现较轻。这项研究丰富了我们对 CIPA 临床和遗传方面的认识,突出了不同的表型和疾病严重程度。
Clinical and genetic characteristics of three patients with congenital insensitivity to pain with anhidrosis: Case reports and a review of the literature
BackgroundCongenital insensitivity to pain with anhidrosis (CIPA) is an extremely rare autosomal recessive disorder caused by loss‐of‐function mutations of the NTRK1 gene, affecting the autonomic and sensory nervous system. Clinical manifestation is varied and includes recurrent fever, pain insensitivity, anhidrosis, self‐mutilating behavior, and intellectual disability.MethodsClinical and genetic features were assessed in two males and one female with genetically confirmed CIPA using exome or genome sequencing.ResultsCIPA symptoms including recurrent fever, pain insensitivity, and anhidrosis manifested at the age of 1 year (age range: 0.3–8 years). Two patients exhibited self‐mutilation tendencies, intellectual disability, and developmental delay. Four NTRK1 (NM_002529.3) mutations, c.851‐33T>A (p.?), c.2020G>T (p.Asp674Tyr), c.2303C>T (p.Pro768Leu), and c.574‐156_850+1113del (exons 5‐7 del) were identified. Two patients exhibited early onset and severe phenotype, being homozygous for c.851‐33T>A (p.?) mutations and compound heterozygous for c.851‐33T>A (p.?) and c.2020G>T (p.Asp674Tyr) mutation of NTRK1. The third patient with compound heterozygous mutations of c.2303C>T (p.Pro768Leu) and c.574‐156_850+1113del (exons 5‐7 del) displayed a late onset and milder clinical manifestation.ConclusionAll three patients exhibited variable phenotypes and disease severity. This research enriches our understanding of clinical and genetic aspects of CIPA, highlighting variable phenotypes and disease severity.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
