Juan S Grassano, Ignacio Pickering, Adrian E Roitberg, Mariano C González Lebrero, Dario A Estrin, Jonathan A Semelak
{"title":"评估混合机器学习/经典电位(ML/MM)方法中的嵌入方案。","authors":"Juan S Grassano, Ignacio Pickering, Adrian E Roitberg, Mariano C González Lebrero, Dario A Estrin, Jonathan A Semelak","doi":"10.1021/acs.jcim.4c00478","DOIUrl":null,"url":null,"abstract":"<p><p>Machine learning (ML) methods have reached high accuracy levels for the prediction of in vacuo molecular properties. However, the simulation of large systems solely through ML methods (such as those based on neural network potentials) is still a challenge. In this context, one of the most promising frameworks for integrating ML schemes in the simulation of complex molecular systems are the so-called ML/MM methods. These multiscale approaches combine ML methods with classical force fields (MM), in the same spirit as the successful hybrid quantum mechanics-molecular mechanics methods (QM/MM). The key issue for such ML/MM methods is an adequate description of the coupling between the region of the system described by ML and the region described at the MM level. In the context of QM/MM schemes, the main ingredient of the interaction is electrostatic, and the state of the art is the so-called electrostatic-embedding. In this study, we analyze the quality of simpler mechanical embedding-based approaches, specifically focusing on their application within a ML/MM framework utilizing atomic partial charges derived in vacuo. Taking as reference electrostatic embedding calculations performed at a QM(DFT)/MM level, we explore different atomic charges schemes, as well as a polarization correction computed using atomic polarizabilites. Our benchmark data set comprises a set of about 80k small organic structures from the ANI-1x and ANI-2x databases, solvated in water. The results suggest that the minimal basis iterative stockholder (MBIS) atomic charges yield the best agreement with the reference coupling energy. Remarkable enhancements are achieved by including a simple polarization correction.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"4047-4058"},"PeriodicalIF":6.4000,"publicationDate":"2024-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Assessment of Embedding Schemes in a Hybrid Machine Learning/Classical Potentials (ML/MM) Approach.\",\"authors\":\"Juan S Grassano, Ignacio Pickering, Adrian E Roitberg, Mariano C González Lebrero, Dario A Estrin, Jonathan A Semelak\",\"doi\":\"10.1021/acs.jcim.4c00478\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Machine learning (ML) methods have reached high accuracy levels for the prediction of in vacuo molecular properties. However, the simulation of large systems solely through ML methods (such as those based on neural network potentials) is still a challenge. In this context, one of the most promising frameworks for integrating ML schemes in the simulation of complex molecular systems are the so-called ML/MM methods. These multiscale approaches combine ML methods with classical force fields (MM), in the same spirit as the successful hybrid quantum mechanics-molecular mechanics methods (QM/MM). The key issue for such ML/MM methods is an adequate description of the coupling between the region of the system described by ML and the region described at the MM level. In the context of QM/MM schemes, the main ingredient of the interaction is electrostatic, and the state of the art is the so-called electrostatic-embedding. In this study, we analyze the quality of simpler mechanical embedding-based approaches, specifically focusing on their application within a ML/MM framework utilizing atomic partial charges derived in vacuo. Taking as reference electrostatic embedding calculations performed at a QM(DFT)/MM level, we explore different atomic charges schemes, as well as a polarization correction computed using atomic polarizabilites. Our benchmark data set comprises a set of about 80k small organic structures from the ANI-1x and ANI-2x databases, solvated in water. The results suggest that the minimal basis iterative stockholder (MBIS) atomic charges yield the best agreement with the reference coupling energy. Remarkable enhancements are achieved by including a simple polarization correction.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\" \",\"pages\":\"4047-4058\"},\"PeriodicalIF\":6.4000,\"publicationDate\":\"2024-05-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jcim.4c00478\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/5/6 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00478","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/5/6 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
摘要
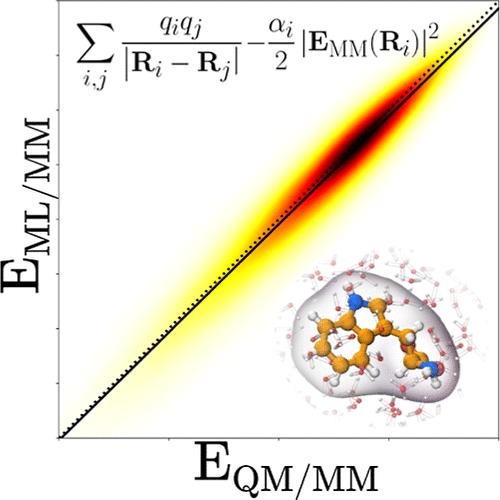
机器学习(ML)方法在预测空泡分子特性方面已经达到了很高的准确度。然而,仅通过 ML 方法(如基于神经网络势能的方法)来模拟大型系统仍然是一项挑战。在这种情况下,将 ML 方案集成到复杂分子系统模拟中的最有前途的框架之一就是所谓的 ML/MM 方法。这些多尺度方法将 ML 方法与经典力场(MM)相结合,其精神与成功的量子力学-分子力学混合方法(QM/MM)相同。这类 ML/MM 方法的关键问题是充分描述 ML 所描述的系统区域与 MM 层面所描述的区域之间的耦合。在 QM/MM 方案中,相互作用的主要成分是静电,而最先进的技术是所谓的静电嵌入。在本研究中,我们分析了较简单的基于机械嵌入的方法的质量,特别关注它们在利用真空中得出的原子偏电荷的 ML/MM 框架中的应用。以在 QM(DFT)/MM 水平上进行的静电嵌入计算为参考,我们探索了不同的原子电荷方案,以及使用原子极化率计算的极化修正。我们的基准数据集包括 ANI-1x 和 ANI-2x 数据库中约 8 万个溶于水的小型有机结构。结果表明,最小基迭代股东(MBIS)原子电荷与参考耦合能的一致性最好。通过加入简单的极化校正,效果显著增强。
Assessment of Embedding Schemes in a Hybrid Machine Learning/Classical Potentials (ML/MM) Approach.
Machine learning (ML) methods have reached high accuracy levels for the prediction of in vacuo molecular properties. However, the simulation of large systems solely through ML methods (such as those based on neural network potentials) is still a challenge. In this context, one of the most promising frameworks for integrating ML schemes in the simulation of complex molecular systems are the so-called ML/MM methods. These multiscale approaches combine ML methods with classical force fields (MM), in the same spirit as the successful hybrid quantum mechanics-molecular mechanics methods (QM/MM). The key issue for such ML/MM methods is an adequate description of the coupling between the region of the system described by ML and the region described at the MM level. In the context of QM/MM schemes, the main ingredient of the interaction is electrostatic, and the state of the art is the so-called electrostatic-embedding. In this study, we analyze the quality of simpler mechanical embedding-based approaches, specifically focusing on their application within a ML/MM framework utilizing atomic partial charges derived in vacuo. Taking as reference electrostatic embedding calculations performed at a QM(DFT)/MM level, we explore different atomic charges schemes, as well as a polarization correction computed using atomic polarizabilites. Our benchmark data set comprises a set of about 80k small organic structures from the ANI-1x and ANI-2x databases, solvated in water. The results suggest that the minimal basis iterative stockholder (MBIS) atomic charges yield the best agreement with the reference coupling energy. Remarkable enhancements are achieved by including a simple polarization correction.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
