E. V. Anikina, D. V. Babailova, M. S. Zhilin, V. P. Beskachko
{"title":"作为储氢材料的四氧杂[8]环烯单层:密度泛函理论中的 Boys-Bernardi 校正模型","authors":"E. V. Anikina, D. V. Babailova, M. S. Zhilin, V. P. Beskachko","doi":"10.1134/S102745102401004X","DOIUrl":null,"url":null,"abstract":"<p>The parameters of molecular hydrogen adsorption on a tetraoxa[8]circulene monolayer were studied using the density functional theory with dispersion interaction corrections (semi-empirical and analytical). The calculations were carried out using two different approaches to the system wave function representation: atomic-like orbital basis set and plane wave basis. Utilizing a less computationally expensive pseudo-atomic basis, it is possible to obtain results for molecular hydrogen adsorption consistent with values calculated with plane waves if the atomic-like basis is optimized and basis set superposition error is corrected for both hydrogen binding energy and geometrical characteristics. Otherwise, the H<sub>2</sub> binding energy will be overestimated by 4–6 times (sometimes even more, by 20); and the hydrogen–monolayer distance will be underestimated by 10–20%. The obtained optimized parameters of the pseudo-atomic basis set can be used for further study of the modified forms of the tetraoxa[8]circulene monolayer. Moreover, our calculations showed that the hydrogen binding to a pristine tetraoxa[8]circulene monolayer is predominantly van der Waals with an energy of 60–90 meV, which is several times less than the desired range of 200–600 meV. To achieve such values, it will be necessary to modify the surface of the monolayer, creating more active sorption cites, for example, by decorating it with metals or applying structural defects.</p>","PeriodicalId":671,"journal":{"name":"Journal of Surface Investigation: X-ray, Synchrotron and Neutron Techniques","volume":"18 1","pages":"19 - 26"},"PeriodicalIF":0.4000,"publicationDate":"2024-05-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Tetraoxa[8]circulene Monolayer as Hydrogen Storage Material: Model with Boys–Bernardi Corrections Within Density Functional Theory\",\"authors\":\"E. V. Anikina, D. V. Babailova, M. S. Zhilin, V. P. Beskachko\",\"doi\":\"10.1134/S102745102401004X\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The parameters of molecular hydrogen adsorption on a tetraoxa[8]circulene monolayer were studied using the density functional theory with dispersion interaction corrections (semi-empirical and analytical). The calculations were carried out using two different approaches to the system wave function representation: atomic-like orbital basis set and plane wave basis. Utilizing a less computationally expensive pseudo-atomic basis, it is possible to obtain results for molecular hydrogen adsorption consistent with values calculated with plane waves if the atomic-like basis is optimized and basis set superposition error is corrected for both hydrogen binding energy and geometrical characteristics. Otherwise, the H<sub>2</sub> binding energy will be overestimated by 4–6 times (sometimes even more, by 20); and the hydrogen–monolayer distance will be underestimated by 10–20%. The obtained optimized parameters of the pseudo-atomic basis set can be used for further study of the modified forms of the tetraoxa[8]circulene monolayer. Moreover, our calculations showed that the hydrogen binding to a pristine tetraoxa[8]circulene monolayer is predominantly van der Waals with an energy of 60–90 meV, which is several times less than the desired range of 200–600 meV. To achieve such values, it will be necessary to modify the surface of the monolayer, creating more active sorption cites, for example, by decorating it with metals or applying structural defects.</p>\",\"PeriodicalId\":671,\"journal\":{\"name\":\"Journal of Surface Investigation: X-ray, Synchrotron and Neutron Techniques\",\"volume\":\"18 1\",\"pages\":\"19 - 26\"},\"PeriodicalIF\":0.4000,\"publicationDate\":\"2024-05-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Surface Investigation: X-ray, Synchrotron and Neutron Techniques\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://link.springer.com/article/10.1134/S102745102401004X\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"PHYSICS, CONDENSED MATTER\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Surface Investigation: X-ray, Synchrotron and Neutron Techniques","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1134/S102745102401004X","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"PHYSICS, CONDENSED MATTER","Score":null,"Total":0}
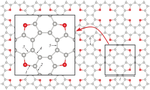
Tetraoxa[8]circulene Monolayer as Hydrogen Storage Material: Model with Boys–Bernardi Corrections Within Density Functional Theory
The parameters of molecular hydrogen adsorption on a tetraoxa[8]circulene monolayer were studied using the density functional theory with dispersion interaction corrections (semi-empirical and analytical). The calculations were carried out using two different approaches to the system wave function representation: atomic-like orbital basis set and plane wave basis. Utilizing a less computationally expensive pseudo-atomic basis, it is possible to obtain results for molecular hydrogen adsorption consistent with values calculated with plane waves if the atomic-like basis is optimized and basis set superposition error is corrected for both hydrogen binding energy and geometrical characteristics. Otherwise, the H2 binding energy will be overestimated by 4–6 times (sometimes even more, by 20); and the hydrogen–monolayer distance will be underestimated by 10–20%. The obtained optimized parameters of the pseudo-atomic basis set can be used for further study of the modified forms of the tetraoxa[8]circulene monolayer. Moreover, our calculations showed that the hydrogen binding to a pristine tetraoxa[8]circulene monolayer is predominantly van der Waals with an energy of 60–90 meV, which is several times less than the desired range of 200–600 meV. To achieve such values, it will be necessary to modify the surface of the monolayer, creating more active sorption cites, for example, by decorating it with metals or applying structural defects.
期刊介绍:
Journal of Surface Investigation: X-ray, Synchrotron and Neutron Techniques publishes original articles on the topical problems of solid-state physics, materials science, experimental techniques, condensed media, nanostructures, surfaces of thin films, and phase boundaries: geometric and energetical structures of surfaces, the methods of computer simulations; physical and chemical properties and their changes upon radiation and other treatments; the methods of studies of films and surface layers of crystals (XRD, XPS, synchrotron radiation, neutron and electron diffraction, electron microscopic, scanning tunneling microscopic, atomic force microscopic studies, and other methods that provide data on the surfaces and thin films). Articles related to the methods and technics of structure studies are the focus of the journal. The journal accepts manuscripts of regular articles and reviews in English or Russian language from authors of all countries. All manuscripts are peer-reviewed.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
