Till Siebenmorgen, Filipe Menezes, Sabrina Benassou, Erinc Merdivan, Kieran Didi, André Santos Dias Mourão, Radosław Kitel, Pietro Liò, Stefan Kesselheim, Marie Piraud, Fabian J. Theis, Michael Sattler, Grzegorz M. Popowicz
{"title":"MISATO:基于结构发现药物的蛋白质配体机器学习数据集。","authors":"Till Siebenmorgen, Filipe Menezes, Sabrina Benassou, Erinc Merdivan, Kieran Didi, André Santos Dias Mourão, Radosław Kitel, Pietro Liò, Stefan Kesselheim, Marie Piraud, Fabian J. Theis, Michael Sattler, Grzegorz M. Popowicz","doi":"10.1038/s43588-024-00627-2","DOIUrl":null,"url":null,"abstract":"Large language models have greatly enhanced our ability to understand biology and chemistry, yet robust methods for structure-based drug discovery, quantum chemistry and structural biology are still sparse. Precise biomolecule–ligand interaction datasets are urgently needed for large language models. To address this, we present MISATO, a dataset that combines quantum mechanical properties of small molecules and associated molecular dynamics simulations of ~20,000 experimental protein–ligand complexes with extensive validation of experimental data. Starting from the existing experimental structures, semi-empirical quantum mechanics was used to systematically refine these structures. A large collection of molecular dynamics traces of protein–ligand complexes in explicit water is included, accumulating over 170 μs. We give examples of machine learning (ML) baseline models proving an improvement of accuracy by employing our data. An easy entry point for ML experts is provided to enable the next generation of drug discovery artificial intelligence models. MISATO is a database for structure-based drug discovery that combines quantum mechanics data with molecular dynamics simulations on ~20,000 protein–ligand structures. The artificial intelligence models included provide an easy entry point for the machine learning and drug discovery communities.","PeriodicalId":74246,"journal":{"name":"Nature computational science","volume":"4 5","pages":"367-378"},"PeriodicalIF":18.3000,"publicationDate":"2024-05-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s43588-024-00627-2.pdf","citationCount":"0","resultStr":"{\"title\":\"MISATO: machine learning dataset of protein–ligand complexes for structure-based drug discovery\",\"authors\":\"Till Siebenmorgen, Filipe Menezes, Sabrina Benassou, Erinc Merdivan, Kieran Didi, André Santos Dias Mourão, Radosław Kitel, Pietro Liò, Stefan Kesselheim, Marie Piraud, Fabian J. Theis, Michael Sattler, Grzegorz M. Popowicz\",\"doi\":\"10.1038/s43588-024-00627-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Large language models have greatly enhanced our ability to understand biology and chemistry, yet robust methods for structure-based drug discovery, quantum chemistry and structural biology are still sparse. Precise biomolecule–ligand interaction datasets are urgently needed for large language models. To address this, we present MISATO, a dataset that combines quantum mechanical properties of small molecules and associated molecular dynamics simulations of ~20,000 experimental protein–ligand complexes with extensive validation of experimental data. Starting from the existing experimental structures, semi-empirical quantum mechanics was used to systematically refine these structures. A large collection of molecular dynamics traces of protein–ligand complexes in explicit water is included, accumulating over 170 μs. We give examples of machine learning (ML) baseline models proving an improvement of accuracy by employing our data. An easy entry point for ML experts is provided to enable the next generation of drug discovery artificial intelligence models. MISATO is a database for structure-based drug discovery that combines quantum mechanics data with molecular dynamics simulations on ~20,000 protein–ligand structures. The artificial intelligence models included provide an easy entry point for the machine learning and drug discovery communities.\",\"PeriodicalId\":74246,\"journal\":{\"name\":\"Nature computational science\",\"volume\":\"4 5\",\"pages\":\"367-378\"},\"PeriodicalIF\":18.3000,\"publicationDate\":\"2024-05-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s43588-024-00627-2.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature computational science\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.nature.com/articles/s43588-024-00627-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature computational science","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s43588-024-00627-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
MISATO: machine learning dataset of protein–ligand complexes for structure-based drug discovery
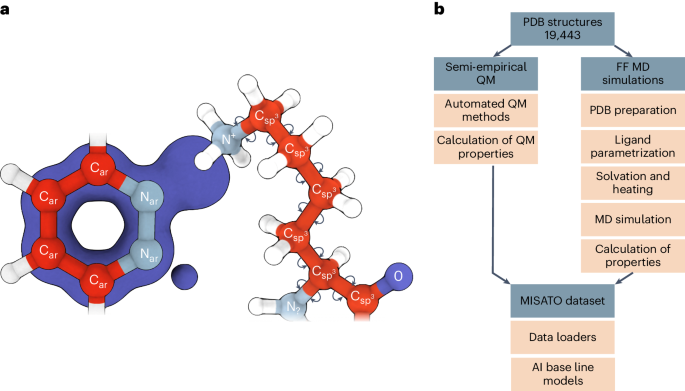
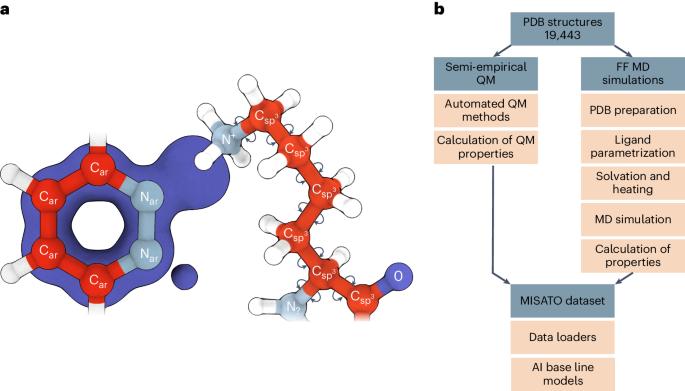
Large language models have greatly enhanced our ability to understand biology and chemistry, yet robust methods for structure-based drug discovery, quantum chemistry and structural biology are still sparse. Precise biomolecule–ligand interaction datasets are urgently needed for large language models. To address this, we present MISATO, a dataset that combines quantum mechanical properties of small molecules and associated molecular dynamics simulations of ~20,000 experimental protein–ligand complexes with extensive validation of experimental data. Starting from the existing experimental structures, semi-empirical quantum mechanics was used to systematically refine these structures. A large collection of molecular dynamics traces of protein–ligand complexes in explicit water is included, accumulating over 170 μs. We give examples of machine learning (ML) baseline models proving an improvement of accuracy by employing our data. An easy entry point for ML experts is provided to enable the next generation of drug discovery artificial intelligence models. MISATO is a database for structure-based drug discovery that combines quantum mechanics data with molecular dynamics simulations on ~20,000 protein–ligand structures. The artificial intelligence models included provide an easy entry point for the machine learning and drug discovery communities.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
