{"title":"取代对分子流动性的限制:1,10-二氰基丁戊烯的案例研究。","authors":"Bin-Bin Pei, Hongjuan Yang, Cai-Yue Gao, Yuan Man, Yonggang Yang, Si-Dian Li","doi":"10.1002/jcc.27379","DOIUrl":null,"url":null,"abstract":"<p>We show herein that 1,10-dicyano substitution restricts the paragon fluxionality of bullvalene to just 14 isomers which isomerize along a single cycle. The restricted fluxionality of 1,10-dicyanobullvalene (DCB) is investigated by means of: (i) Bonding analyses of the isomer structures using the adaptive natural density partitioning (AdNDP). (ii) Quantum dynamical simulations of the isomerizations along the cyclic intrinsic reaction coordinate of the potential energy surface (PES). The PES possesses 14 equivalent potential wells supporting 14 isomers which are separated by 14 equivalent potential barriers supporting 14 transition states. Accordingly, at low temperatures, DCB appears as a hindered molecular rotor, without any delocalization of the wavefunction in the 14 potential wells, without any nuclear spin isomers, and with completely negligible tunneling. These results are compared and found to differ from those for molecular boron rotors. (iii) Born-Oppenheimer molecular dynamics (BOMD) simulations of thermally activated isomerizations. (iv) Calculations of the rate constants in the frame of transition state theory (TST) with reasonable agreement achieved with the BOMD results. (v) Simulations of the equilibration dynamics using rate equations for the isomerizations with TST rate coefficients. Accordingly, in the long-time limit, isomerizations of the 14 isomers, each with C<sub><i>s</i></sub> symmetry, approach the “14 C<sub><i>s</i></sub> → C<sub>7<i>v</i></sub>” thermally averaged structure. This is a superposition of the 14 equally populated isomer structures with an overall C<sub>7<i>v</i></sub> symmetry. By extrapolation, the results for DCB yield working hypotheses for so far un-explored properties e.g. for the equilibration dynamics of C<sub>10</sub>H<sub>10</sub>.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 24","pages":"2080-2090"},"PeriodicalIF":3.4000,"publicationDate":"2024-05-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Restriction on molecular fluxionality by substitution: A case study for the 1,10-dicyanobullvalene\",\"authors\":\"Bin-Bin Pei, Hongjuan Yang, Cai-Yue Gao, Yuan Man, Yonggang Yang, Si-Dian Li\",\"doi\":\"10.1002/jcc.27379\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>We show herein that 1,10-dicyano substitution restricts the paragon fluxionality of bullvalene to just 14 isomers which isomerize along a single cycle. The restricted fluxionality of 1,10-dicyanobullvalene (DCB) is investigated by means of: (i) Bonding analyses of the isomer structures using the adaptive natural density partitioning (AdNDP). (ii) Quantum dynamical simulations of the isomerizations along the cyclic intrinsic reaction coordinate of the potential energy surface (PES). The PES possesses 14 equivalent potential wells supporting 14 isomers which are separated by 14 equivalent potential barriers supporting 14 transition states. Accordingly, at low temperatures, DCB appears as a hindered molecular rotor, without any delocalization of the wavefunction in the 14 potential wells, without any nuclear spin isomers, and with completely negligible tunneling. These results are compared and found to differ from those for molecular boron rotors. (iii) Born-Oppenheimer molecular dynamics (BOMD) simulations of thermally activated isomerizations. (iv) Calculations of the rate constants in the frame of transition state theory (TST) with reasonable agreement achieved with the BOMD results. (v) Simulations of the equilibration dynamics using rate equations for the isomerizations with TST rate coefficients. Accordingly, in the long-time limit, isomerizations of the 14 isomers, each with C<sub><i>s</i></sub> symmetry, approach the “14 C<sub><i>s</i></sub> → C<sub>7<i>v</i></sub>” thermally averaged structure. This is a superposition of the 14 equally populated isomer structures with an overall C<sub>7<i>v</i></sub> symmetry. By extrapolation, the results for DCB yield working hypotheses for so far un-explored properties e.g. for the equilibration dynamics of C<sub>10</sub>H<sub>10</sub>.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 24\",\"pages\":\"2080-2090\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-05-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27379\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27379","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Restriction on molecular fluxionality by substitution: A case study for the 1,10-dicyanobullvalene
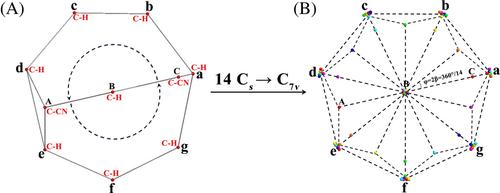
We show herein that 1,10-dicyano substitution restricts the paragon fluxionality of bullvalene to just 14 isomers which isomerize along a single cycle. The restricted fluxionality of 1,10-dicyanobullvalene (DCB) is investigated by means of: (i) Bonding analyses of the isomer structures using the adaptive natural density partitioning (AdNDP). (ii) Quantum dynamical simulations of the isomerizations along the cyclic intrinsic reaction coordinate of the potential energy surface (PES). The PES possesses 14 equivalent potential wells supporting 14 isomers which are separated by 14 equivalent potential barriers supporting 14 transition states. Accordingly, at low temperatures, DCB appears as a hindered molecular rotor, without any delocalization of the wavefunction in the 14 potential wells, without any nuclear spin isomers, and with completely negligible tunneling. These results are compared and found to differ from those for molecular boron rotors. (iii) Born-Oppenheimer molecular dynamics (BOMD) simulations of thermally activated isomerizations. (iv) Calculations of the rate constants in the frame of transition state theory (TST) with reasonable agreement achieved with the BOMD results. (v) Simulations of the equilibration dynamics using rate equations for the isomerizations with TST rate coefficients. Accordingly, in the long-time limit, isomerizations of the 14 isomers, each with Cs symmetry, approach the “14 Cs → C7v” thermally averaged structure. This is a superposition of the 14 equally populated isomer structures with an overall C7v symmetry. By extrapolation, the results for DCB yield working hypotheses for so far un-explored properties e.g. for the equilibration dynamics of C10H10.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
