{"title":"分子动力学模拟中的神经网络潜力概述","authors":"Raidel Martin-Barrios, Edisel Navas-Conyedo, Xuyi Zhang, Yunwei Chen, Jorge Gulín-González","doi":"10.1002/qua.27389","DOIUrl":null,"url":null,"abstract":"<p>Ab-initio molecular dynamics (AIMD) is a key method for realistic simulation of complex atomistic systems and processes in nanoscale. In AIMD, finite-temperature dynamical trajectories are generated by using forces computed from electronic structure calculations. In systems with high numbers of components a typical AIMD run is computationally demanding. On the other hand, machine learning (ML) is a subfield of the artificial intelligence that consist in a set of algorithms that show learning by experience with the use of input and output data where algorithms are capable of analysing and predicting the future. At present, the main application of ML techniques in atomic simulations is the development of new interatomic potentials to correctly describe the potential energy surfaces (PES). This technique is in constant progress since its inception around 30 years ago. The ML potentials combine the advantages of classical and Ab-initio methods, that is, the efficiency of a simple functional form and the accuracy of first principles calculations. In this article we review the evolution of four generations of machine learning potentials and some of their most notable applications. This review focuses on MLPs based on neural networks. Also, we present a state of art of this topic and future trends. Finally, we report the results of a scientometric study (covering the period 1995–2023) about the impact of ML techniques applied to atomistic simulations, distribution of publications by geographical regions and hot topics investigated in the literature.</p>","PeriodicalId":182,"journal":{"name":"International Journal of Quantum Chemistry","volume":"124 11","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-05-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/qua.27389","citationCount":"0","resultStr":"{\"title\":\"An overview about neural networks potentials in molecular dynamics simulation\",\"authors\":\"Raidel Martin-Barrios, Edisel Navas-Conyedo, Xuyi Zhang, Yunwei Chen, Jorge Gulín-González\",\"doi\":\"10.1002/qua.27389\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Ab-initio molecular dynamics (AIMD) is a key method for realistic simulation of complex atomistic systems and processes in nanoscale. In AIMD, finite-temperature dynamical trajectories are generated by using forces computed from electronic structure calculations. In systems with high numbers of components a typical AIMD run is computationally demanding. On the other hand, machine learning (ML) is a subfield of the artificial intelligence that consist in a set of algorithms that show learning by experience with the use of input and output data where algorithms are capable of analysing and predicting the future. At present, the main application of ML techniques in atomic simulations is the development of new interatomic potentials to correctly describe the potential energy surfaces (PES). This technique is in constant progress since its inception around 30 years ago. The ML potentials combine the advantages of classical and Ab-initio methods, that is, the efficiency of a simple functional form and the accuracy of first principles calculations. In this article we review the evolution of four generations of machine learning potentials and some of their most notable applications. This review focuses on MLPs based on neural networks. Also, we present a state of art of this topic and future trends. Finally, we report the results of a scientometric study (covering the period 1995–2023) about the impact of ML techniques applied to atomistic simulations, distribution of publications by geographical regions and hot topics investigated in the literature.</p>\",\"PeriodicalId\":182,\"journal\":{\"name\":\"International Journal of Quantum Chemistry\",\"volume\":\"124 11\",\"pages\":\"\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2024-05-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/qua.27389\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Quantum Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/qua.27389\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Quantum Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/qua.27389","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
An overview about neural networks potentials in molecular dynamics simulation
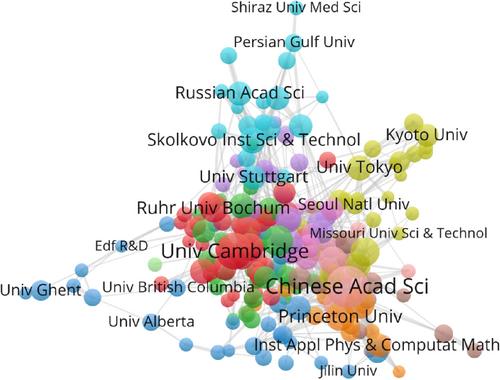
Ab-initio molecular dynamics (AIMD) is a key method for realistic simulation of complex atomistic systems and processes in nanoscale. In AIMD, finite-temperature dynamical trajectories are generated by using forces computed from electronic structure calculations. In systems with high numbers of components a typical AIMD run is computationally demanding. On the other hand, machine learning (ML) is a subfield of the artificial intelligence that consist in a set of algorithms that show learning by experience with the use of input and output data where algorithms are capable of analysing and predicting the future. At present, the main application of ML techniques in atomic simulations is the development of new interatomic potentials to correctly describe the potential energy surfaces (PES). This technique is in constant progress since its inception around 30 years ago. The ML potentials combine the advantages of classical and Ab-initio methods, that is, the efficiency of a simple functional form and the accuracy of first principles calculations. In this article we review the evolution of four generations of machine learning potentials and some of their most notable applications. This review focuses on MLPs based on neural networks. Also, we present a state of art of this topic and future trends. Finally, we report the results of a scientometric study (covering the period 1995–2023) about the impact of ML techniques applied to atomistic simulations, distribution of publications by geographical regions and hot topics investigated in the literature.
期刊介绍:
Since its first formulation quantum chemistry has provided the conceptual and terminological framework necessary to understand atoms, molecules and the condensed matter. Over the past decades synergistic advances in the methodological developments, software and hardware have transformed quantum chemistry in a truly interdisciplinary science that has expanded beyond its traditional core of molecular sciences to fields as diverse as chemistry and catalysis, biophysics, nanotechnology and material science.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
