{"title":"使用 FMO 方案进行 VQE-UCCSD 计算时发现的尺寸一致性和轨道不变性问题。","authors":"Kenji Sugisaki, Tatsuya Nakano, Yuji Mochizuki","doi":"10.1002/jcc.27438","DOIUrl":null,"url":null,"abstract":"<p>The fragment molecular orbital (FMO) scheme is one of the popular fragmentation-based methods and has the potential advantage of making the circuit shallow for quantum chemical calculations on quantum computers. In this study, we used a GPU-accelerated quantum simulator (cuQuantum) to perform the electron correlation part of the FMO calculation as unitary coupled-cluster singles and doubles (UCCSD) with the variational quantum eigensolver (VQE) for hydrogen-bonded (FH)<span></span><math>\n <mrow>\n <msub>\n <mo> </mo>\n <mrow>\n <mn>3</mn>\n </mrow>\n </msub>\n </mrow></math> and (FH)<span></span><math>\n <mrow>\n <msub>\n <mo> </mo>\n <mrow>\n <mn>2</mn>\n </mrow>\n </msub>\n </mrow></math>-H<span></span><math>\n <mrow>\n <msub>\n <mo> </mo>\n <mrow>\n <mn>2</mn>\n </mrow>\n </msub>\n </mrow></math>O systems with the STO-3G basis set. VQE-UCCSD calculations were performed using both canonical and localized MO sets, and the results were examined from the point of view of size-consistency and orbital-invariance affected by the Trotter error. It was found that the use of localized MO leads to better results, especially for (FH)<span></span><math>\n <mrow>\n <msub>\n <mo> </mo>\n <mrow>\n <mn>2</mn>\n </mrow>\n </msub>\n </mrow></math>-H<span></span><math>\n <mrow>\n <msub>\n <mo> </mo>\n <mrow>\n <mn>2</mn>\n </mrow>\n </msub>\n </mrow></math>O. The GPU acceleration was substantial for the simulations with larger numbers of qubits, and was about a factor of 6.7–7.7 for 18 qubit systems.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 26","pages":"2204-2213"},"PeriodicalIF":2.9000,"publicationDate":"2024-05-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27438","citationCount":"0","resultStr":"{\"title\":\"Size-consistency and orbital-invariance issues revealed by VQE-UCCSD calculations with the FMO scheme\",\"authors\":\"Kenji Sugisaki, Tatsuya Nakano, Yuji Mochizuki\",\"doi\":\"10.1002/jcc.27438\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The fragment molecular orbital (FMO) scheme is one of the popular fragmentation-based methods and has the potential advantage of making the circuit shallow for quantum chemical calculations on quantum computers. In this study, we used a GPU-accelerated quantum simulator (cuQuantum) to perform the electron correlation part of the FMO calculation as unitary coupled-cluster singles and doubles (UCCSD) with the variational quantum eigensolver (VQE) for hydrogen-bonded (FH)<span></span><math>\\n <mrow>\\n <msub>\\n <mo> </mo>\\n <mrow>\\n <mn>3</mn>\\n </mrow>\\n </msub>\\n </mrow></math> and (FH)<span></span><math>\\n <mrow>\\n <msub>\\n <mo> </mo>\\n <mrow>\\n <mn>2</mn>\\n </mrow>\\n </msub>\\n </mrow></math>-H<span></span><math>\\n <mrow>\\n <msub>\\n <mo> </mo>\\n <mrow>\\n <mn>2</mn>\\n </mrow>\\n </msub>\\n </mrow></math>O systems with the STO-3G basis set. VQE-UCCSD calculations were performed using both canonical and localized MO sets, and the results were examined from the point of view of size-consistency and orbital-invariance affected by the Trotter error. It was found that the use of localized MO leads to better results, especially for (FH)<span></span><math>\\n <mrow>\\n <msub>\\n <mo> </mo>\\n <mrow>\\n <mn>2</mn>\\n </mrow>\\n </msub>\\n </mrow></math>-H<span></span><math>\\n <mrow>\\n <msub>\\n <mo> </mo>\\n <mrow>\\n <mn>2</mn>\\n </mrow>\\n </msub>\\n </mrow></math>O. The GPU acceleration was substantial for the simulations with larger numbers of qubits, and was about a factor of 6.7–7.7 for 18 qubit systems.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 26\",\"pages\":\"2204-2213\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-05-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27438\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27438\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27438","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Size-consistency and orbital-invariance issues revealed by VQE-UCCSD calculations with the FMO scheme
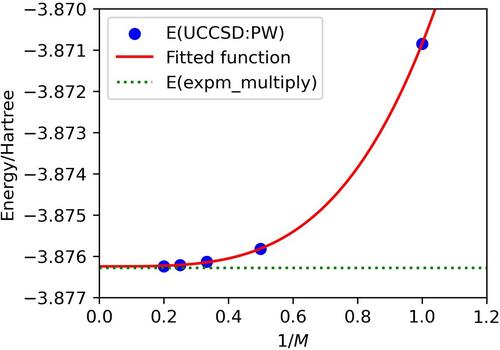
The fragment molecular orbital (FMO) scheme is one of the popular fragmentation-based methods and has the potential advantage of making the circuit shallow for quantum chemical calculations on quantum computers. In this study, we used a GPU-accelerated quantum simulator (cuQuantum) to perform the electron correlation part of the FMO calculation as unitary coupled-cluster singles and doubles (UCCSD) with the variational quantum eigensolver (VQE) for hydrogen-bonded (FH) and (FH)-HO systems with the STO-3G basis set. VQE-UCCSD calculations were performed using both canonical and localized MO sets, and the results were examined from the point of view of size-consistency and orbital-invariance affected by the Trotter error. It was found that the use of localized MO leads to better results, especially for (FH)-HO. The GPU acceleration was substantial for the simulations with larger numbers of qubits, and was about a factor of 6.7–7.7 for 18 qubit systems.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
