Arnab Jana, Sam Shepherd, Yair Litman and David M. Wilkins*,
{"title":"学习水体系中的电子极化。","authors":"Arnab Jana, Sam Shepherd, Yair Litman and David M. Wilkins*, ","doi":"10.1021/acs.jcim.4c00421","DOIUrl":null,"url":null,"abstract":"<p >The polarization of periodically repeating systems is a discontinuous function of the atomic positions, a fact which seems at first to stymie attempts at their statistical learning. Two approaches to build models for bulk polarizations are compared: one in which a simple point charge model is used to preprocess the raw polarization to give a learning target that is a smooth function of atomic positions and the total polarization is learned as a sum of atom-centered dipoles and one in which instead the average position of Wannier centers around atoms is predicted. For a range of bulk aqueous systems, both of these methods perform perform comparatively well, with the former being slightly better but often requiring an extra effort to find a suitable point charge model. As a challenging test, we also analyze the performance of the models at the air–water interface. In this case, while the Wannier center approach delivers accurate predictions without further modifications, the preprocessing method requires augmentation with information from isolated water molecules to reach similar accuracy. Finally, we present a simple protocol to preprocess the polarizations in a data-driven way using a small number of derivatives calculated at a much lower level of theory, thus overcoming the need to find point charge models without appreciably increasing the computation cost. We believe that the training strategies presented here help the construction of accurate polarization models required for the study of the dielectric properties of realistic complex bulk systems and interfaces with ab initio accuracy.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 11","pages":"4426–4435"},"PeriodicalIF":6.4000,"publicationDate":"2024-05-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jcim.4c00421","citationCount":"0","resultStr":"{\"title\":\"Learning Electronic Polarizations in Aqueous Systems\",\"authors\":\"Arnab Jana, Sam Shepherd, Yair Litman and David M. Wilkins*, \",\"doi\":\"10.1021/acs.jcim.4c00421\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The polarization of periodically repeating systems is a discontinuous function of the atomic positions, a fact which seems at first to stymie attempts at their statistical learning. Two approaches to build models for bulk polarizations are compared: one in which a simple point charge model is used to preprocess the raw polarization to give a learning target that is a smooth function of atomic positions and the total polarization is learned as a sum of atom-centered dipoles and one in which instead the average position of Wannier centers around atoms is predicted. For a range of bulk aqueous systems, both of these methods perform perform comparatively well, with the former being slightly better but often requiring an extra effort to find a suitable point charge model. As a challenging test, we also analyze the performance of the models at the air–water interface. In this case, while the Wannier center approach delivers accurate predictions without further modifications, the preprocessing method requires augmentation with information from isolated water molecules to reach similar accuracy. Finally, we present a simple protocol to preprocess the polarizations in a data-driven way using a small number of derivatives calculated at a much lower level of theory, thus overcoming the need to find point charge models without appreciably increasing the computation cost. We believe that the training strategies presented here help the construction of accurate polarization models required for the study of the dielectric properties of realistic complex bulk systems and interfaces with ab initio accuracy.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"64 11\",\"pages\":\"4426–4435\"},\"PeriodicalIF\":6.4000,\"publicationDate\":\"2024-05-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.jcim.4c00421\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00421\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00421","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Learning Electronic Polarizations in Aqueous Systems
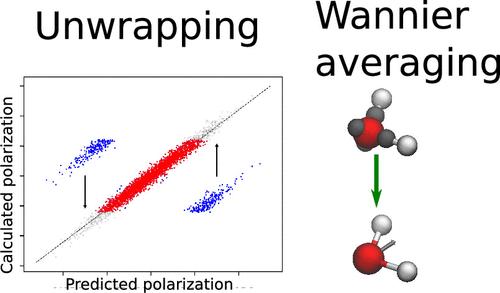
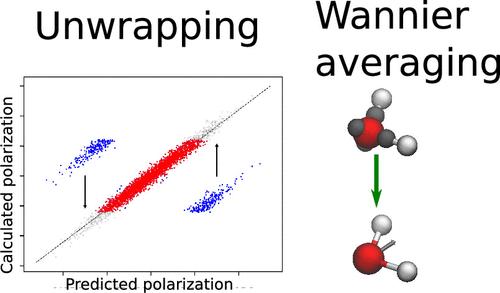
The polarization of periodically repeating systems is a discontinuous function of the atomic positions, a fact which seems at first to stymie attempts at their statistical learning. Two approaches to build models for bulk polarizations are compared: one in which a simple point charge model is used to preprocess the raw polarization to give a learning target that is a smooth function of atomic positions and the total polarization is learned as a sum of atom-centered dipoles and one in which instead the average position of Wannier centers around atoms is predicted. For a range of bulk aqueous systems, both of these methods perform perform comparatively well, with the former being slightly better but often requiring an extra effort to find a suitable point charge model. As a challenging test, we also analyze the performance of the models at the air–water interface. In this case, while the Wannier center approach delivers accurate predictions without further modifications, the preprocessing method requires augmentation with information from isolated water molecules to reach similar accuracy. Finally, we present a simple protocol to preprocess the polarizations in a data-driven way using a small number of derivatives calculated at a much lower level of theory, thus overcoming the need to find point charge models without appreciably increasing the computation cost. We believe that the training strategies presented here help the construction of accurate polarization models required for the study of the dielectric properties of realistic complex bulk systems and interfaces with ab initio accuracy.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
