{"title":"硅、掺 N 改性石墨烯量子点支撑单原子铁催化 CO2 加氢制甲酸的理论研究","authors":"Fangfang Li, Xunchao Zhang, Lihua Kang","doi":"10.1002/qua.27425","DOIUrl":null,"url":null,"abstract":"<p>Exploring suitable catalysts to catalyze the chemical transformation of CO<sub>2</sub> molecules is essential to reduce CO<sub>2</sub> levels. In this article, catalyst models of Fe-C<sub>4</sub>, Fe-N<sub>4</sub>, and Fe-Si<sub>4</sub> were constructed using the density functional theory (DFT) calculations, and the reaction mechanisms of CO<sub>2</sub> hydrogenation over these three catalysts were calculated and analyzed. The results showed that the doping of N atoms lowered the energy barrier of the second hydrogenation step compared with that of Fe-C<sub>4</sub> catalyst, while the doping of Si atoms changed the electron distribution on the surface of the catalyst and formed new Si adsorption sites. And the Fe-Si<sub>4</sub> catalyst had a stronger ability to activate CO<sub>2</sub> molecules as well as stronger catalytic performance compared with the Fe-C<sub>4</sub> and Fe-N<sub>4</sub> catalysts, which was mainly attributed to the synergistic catalytic effect between the doped Si atoms and the Fe metal atom.</p>","PeriodicalId":182,"journal":{"name":"International Journal of Quantum Chemistry","volume":"124 11","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-05-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Theory study on catalytic hydrogenation of CO2 to formic acid over Si, N-doped modified graphene quantum dots supported single atom Fe\",\"authors\":\"Fangfang Li, Xunchao Zhang, Lihua Kang\",\"doi\":\"10.1002/qua.27425\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Exploring suitable catalysts to catalyze the chemical transformation of CO<sub>2</sub> molecules is essential to reduce CO<sub>2</sub> levels. In this article, catalyst models of Fe-C<sub>4</sub>, Fe-N<sub>4</sub>, and Fe-Si<sub>4</sub> were constructed using the density functional theory (DFT) calculations, and the reaction mechanisms of CO<sub>2</sub> hydrogenation over these three catalysts were calculated and analyzed. The results showed that the doping of N atoms lowered the energy barrier of the second hydrogenation step compared with that of Fe-C<sub>4</sub> catalyst, while the doping of Si atoms changed the electron distribution on the surface of the catalyst and formed new Si adsorption sites. And the Fe-Si<sub>4</sub> catalyst had a stronger ability to activate CO<sub>2</sub> molecules as well as stronger catalytic performance compared with the Fe-C<sub>4</sub> and Fe-N<sub>4</sub> catalysts, which was mainly attributed to the synergistic catalytic effect between the doped Si atoms and the Fe metal atom.</p>\",\"PeriodicalId\":182,\"journal\":{\"name\":\"International Journal of Quantum Chemistry\",\"volume\":\"124 11\",\"pages\":\"\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2024-05-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Quantum Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/qua.27425\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Quantum Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/qua.27425","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
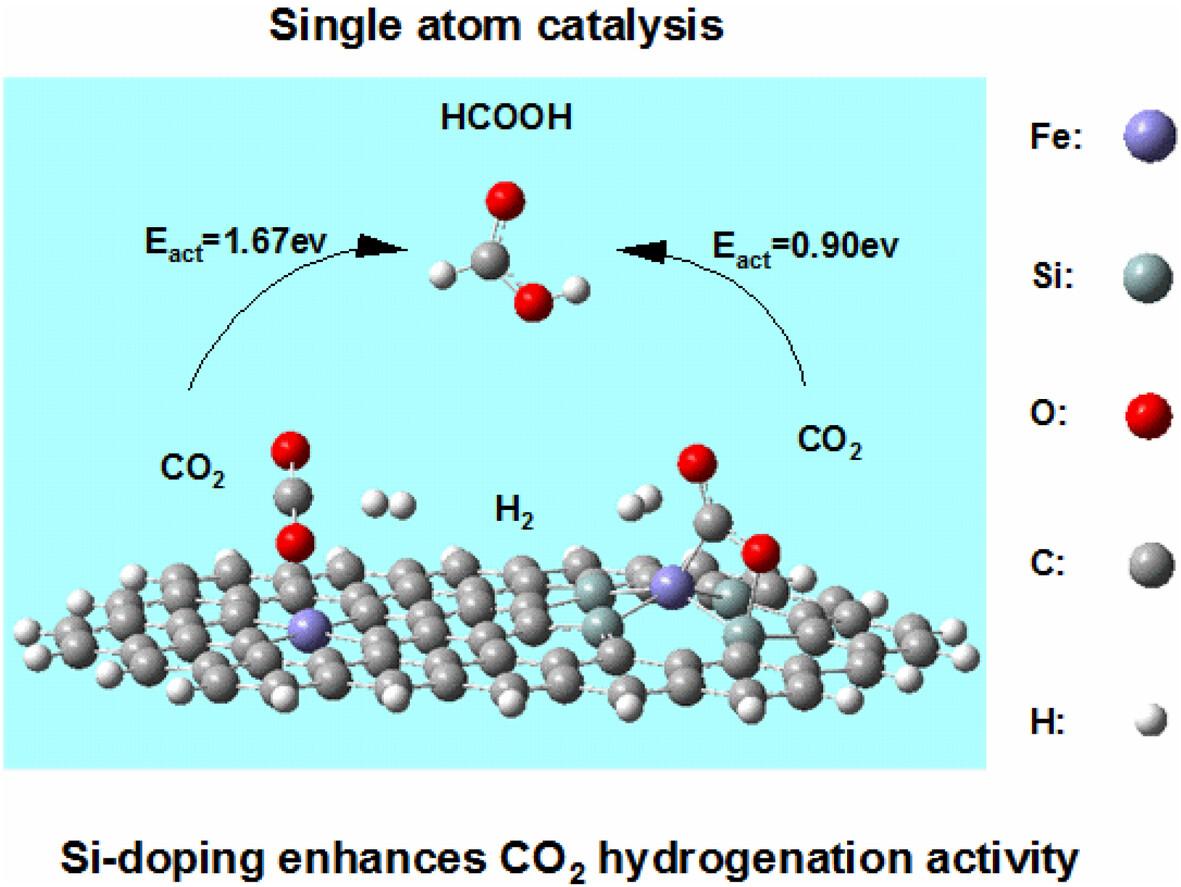
探索合适的催化剂来催化二氧化碳分子的化学转化对于降低二氧化碳含量至关重要。本文利用密度泛函理论(DFT)计算,构建了Fe-C4、Fe-N4和Fe-Si4催化剂模型,并计算分析了这三种催化剂的二氧化碳加氢反应机理。结果表明,与 Fe-C4 催化剂相比,N 原子的掺杂降低了第二步加氢的能垒;而 Si 原子的掺杂改变了催化剂表面的电子分布,形成了新的 Si 吸附位点。与 Fe-C4 和 Fe-N4 催化剂相比,Fe-Si4 催化剂活化 CO2 分子的能力更强,催化性能也更高,这主要归功于掺杂的 Si 原子与 Fe 金属原子之间的协同催化作用。
Theory study on catalytic hydrogenation of CO2 to formic acid over Si, N-doped modified graphene quantum dots supported single atom Fe
Exploring suitable catalysts to catalyze the chemical transformation of CO2 molecules is essential to reduce CO2 levels. In this article, catalyst models of Fe-C4, Fe-N4, and Fe-Si4 were constructed using the density functional theory (DFT) calculations, and the reaction mechanisms of CO2 hydrogenation over these three catalysts were calculated and analyzed. The results showed that the doping of N atoms lowered the energy barrier of the second hydrogenation step compared with that of Fe-C4 catalyst, while the doping of Si atoms changed the electron distribution on the surface of the catalyst and formed new Si adsorption sites. And the Fe-Si4 catalyst had a stronger ability to activate CO2 molecules as well as stronger catalytic performance compared with the Fe-C4 and Fe-N4 catalysts, which was mainly attributed to the synergistic catalytic effect between the doped Si atoms and the Fe metal atom.
期刊介绍:
Since its first formulation quantum chemistry has provided the conceptual and terminological framework necessary to understand atoms, molecules and the condensed matter. Over the past decades synergistic advances in the methodological developments, software and hardware have transformed quantum chemistry in a truly interdisciplinary science that has expanded beyond its traditional core of molecular sciences to fields as diverse as chemistry and catalysis, biophysics, nanotechnology and material science.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
