{"title":"NPEX:利用深度强化学习永不放弃蛋白质探索","authors":"Yuta Shimono, Masataka Hakamada, Mamoru Mabuchi","doi":"10.1016/j.jmgm.2024.108802","DOIUrl":null,"url":null,"abstract":"<div><p>Elucidating unknown structures of proteins, such as metastable states, is critical in designing therapeutic agents. Protein structure exploration has been performed using advanced computational methods, especially molecular dynamics and Markov chain Monte Carlo simulations, which require untenably long calculation times and prior structural knowledge. Here, we developed an innovative method for protein structure determination called never give up protein exploration (NPEX) with deep reinforcement learning. The NPEX method leverages the soft actor-critic algorithm and the intrinsic reward system, effectively adding a bias potential without the need for prior knowledge. To demonstrate the method's effectiveness, we applied it to four models: a double well, a triple well, the alanine dipeptide, and the tryptophan cage. Compared with Markov chain Monte Carlo simulations, NPEX had markedly greater sampling efficiency. The significantly enhanced computational efficiency and lack of prior domain knowledge requirements of the NPEX method will revolutionize protein structure exploration.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"131 ","pages":"Article 108802"},"PeriodicalIF":3.1000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"NPEX: Never give up protein exploration with deep reinforcement learning\",\"authors\":\"Yuta Shimono, Masataka Hakamada, Mamoru Mabuchi\",\"doi\":\"10.1016/j.jmgm.2024.108802\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Elucidating unknown structures of proteins, such as metastable states, is critical in designing therapeutic agents. Protein structure exploration has been performed using advanced computational methods, especially molecular dynamics and Markov chain Monte Carlo simulations, which require untenably long calculation times and prior structural knowledge. Here, we developed an innovative method for protein structure determination called never give up protein exploration (NPEX) with deep reinforcement learning. The NPEX method leverages the soft actor-critic algorithm and the intrinsic reward system, effectively adding a bias potential without the need for prior knowledge. To demonstrate the method's effectiveness, we applied it to four models: a double well, a triple well, the alanine dipeptide, and the tryptophan cage. Compared with Markov chain Monte Carlo simulations, NPEX had markedly greater sampling efficiency. The significantly enhanced computational efficiency and lack of prior domain knowledge requirements of the NPEX method will revolutionize protein structure exploration.</p></div>\",\"PeriodicalId\":16361,\"journal\":{\"name\":\"Journal of molecular graphics & modelling\",\"volume\":\"131 \",\"pages\":\"Article 108802\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular graphics & modelling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1093326324001025\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/5/25 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001025","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/5/25 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
NPEX: Never give up protein exploration with deep reinforcement learning
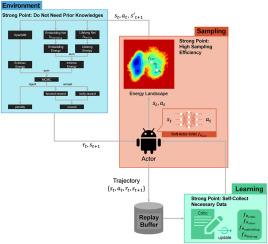
Elucidating unknown structures of proteins, such as metastable states, is critical in designing therapeutic agents. Protein structure exploration has been performed using advanced computational methods, especially molecular dynamics and Markov chain Monte Carlo simulations, which require untenably long calculation times and prior structural knowledge. Here, we developed an innovative method for protein structure determination called never give up protein exploration (NPEX) with deep reinforcement learning. The NPEX method leverages the soft actor-critic algorithm and the intrinsic reward system, effectively adding a bias potential without the need for prior knowledge. To demonstrate the method's effectiveness, we applied it to four models: a double well, a triple well, the alanine dipeptide, and the tryptophan cage. Compared with Markov chain Monte Carlo simulations, NPEX had markedly greater sampling efficiency. The significantly enhanced computational efficiency and lack of prior domain knowledge requirements of the NPEX method will revolutionize protein structure exploration.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
