Fei Gao, Shuang Chen, Jianwei Li, Zailin Yang, Cui Mao
{"title":"植物血凝素刺激的细胞培养在 T 细胞原淋巴细胞白血病诊断中的关键作用:基于病例的方法。","authors":"Fei Gao, Shuang Chen, Jianwei Li, Zailin Yang, Cui Mao","doi":"10.1111/ijlh.14323","DOIUrl":null,"url":null,"abstract":"<p>T-cell prolymphocytic leukemia (T-PLL) is a very rare subtype of the mature lymphocytic malignancies that typically occur in middle-aged and older individuals, accounting for approximately 2% of all mature T-cell lymphomas.<span><sup>1</sup></span> T-PLL is characterized by a poor median survival rate and distinctive cell morphological, immunophenotypic, and cytogenetic features. The major diagnostic criteria of the guidelines for T-PLL diagnosis<span><sup>2</sup></span> include: (1) 5 × 10<sup>9</sup>/L cells of the T-PLL phenotype in peripheral blood or bone marrow (BM); (2) T-cell clonality determined via PCR or flow cytometry (FCM); and, (3) an abnormal 14q32 or Xq28 karyotype or expression of <i>TCL1A/B</i> or <i>MTCP1</i>. Additionally, four minor criteria also contribute to the diagnostic framework: (1) abnormalities involving chromosome 11 (11q22.3; <i>ATM</i>); (2) abnormalities involving chromosome 8 such as idic(8)(p11), t(8;8), trisomy 8q; (3) abnormalities in chromosomes 5, 12, 13, 22, or a complex karyotype; and, (4) T-PLL-specific features (e.g., splenomegaly, effusions). A diagnosis of T-PLL is established if all three major criteria are met or if the first two such criteria and at least one minor criterion are met. Obviously, analysis of chromosomal abnormalities is essential for diagnosis of T-PLL. However, T-PLL poses a challenge in this regard given the low proliferation capacity of mature T lymphocytes, leading to a high failure rate of chromosomal karyotyping associated with conventional culture methods (short-term culture).</p><p>In this study, we retrospectively analyze and present five cases for whom we used a phytohemagglutinin (PHA)-stimulated culture method to increase the detection rate of chromosomal abnormalities in BM cells. This enhanced T-PLL diagnostic accuracy.</p><p>All five patients were males of median age 58 years (range, 42 to 83 years). Immunophenotypic abnormalities were observed in 50% ~ 90% of T cells of all patients (Table S1) in BM sample. Most exhibited the phenotype CD3<sup>+</sup>, CD2<sup>+</sup>, CD4<sup>+</sup>, CD5<sup>+</sup>, CD7<sup>+</sup>, CD56<sup>−</sup>, CD34<sup>−</sup>, CD117<sup>−</sup>, CD1a<sup>−</sup>, and CD52<sup>+</sup>. The most notable immunophenotypic difference was that for CD8: cases 1, 4, and 5 were of CD8<sup>part+</sup> status; case 2 CD8<sup>−</sup>; and case 3 CD8<sup>+</sup>. Subsequently, chromosomal karyotyping was performed on cultures grown with and without PHA (Table 1). Three of the five patients (cases 1, 2, and 3) exhibited only a 46,XY karyotype when cells were grown without PHA, but complex karyotypes, inv(14), trisomy 8q, and abnormalities in 11q when cells were grown with PHA. One patient (case 5) exhibited no metaphase cells when cells were grown without PHA, but a complex karyotype, abnormalities in 11q, and t (14;14) when cells were grown with PHA. Only one patient (case 4) yielded consistent chromosomal karyotypes when cells were grown with and without PHA, thus add(3)(q26), i(8)(q10), inv(14)(q11q32), add(16)(q22), and add(21)(p11). Figure 1 shows the chromosomal karyotypes afforded by the PHA-stimulated cell culture approach for all cases. Obviously, the use of PHA-stimulated BM cell cultures for chromosomal karyotyping significantly increased the detection rates of chromosomal abnormalities (4/5, 80%). PHA, a pan-lymphocyte mitogen, effectively stimulates T cell proliferation and differentiation. Although the properties of PHA-conditioned medium have been described in previous studies,<span><sup>3-5</sup></span> culture of BM cells with PHA is not commonly used in clinical practice for chromosomal karyotyping of patients under suspicion of abnormal, mature T lymphocyte status. Our results further emphasize that PHA-stimulated cell culture enhances the detection of chromosomal abnormalities in T-PLL patients.</p><p>Strikingly, we found that four of the five patients (cases 1, 2, 3, and 4) were of inv(14)(q11q32) status and one (case 5) of t(14;14)(q11;q32) status. Dürig et al. reported that 80% of T-PLL patients exhibited complex chromosomal karyotypes such as inv(14)(q11q32).<span><sup>6</sup></span> The 14q32 abnormality frequently triggers formation of the <i>TRA-TRD::TCL1A</i> fusion gene, a notable genetic aberration of T-PLL.<span><sup>7-9</sup></span> In two of our patients (cases 1, 4), <i>TCL1</i> rearrangements were identified via fluorescence in situ hybridization (FISH) (Figure S1). Furthermore, abnormalities in chromosomes 8 and 12p, and deletions in the long arms of chromosomes 5, 6, 11, and 13, have been reported in T-PLL patients.<span><sup>7</sup></span> Several studies also suggested that 40% ~ 80% of T-PLL patients exhibit trisomy 8q.<span><sup>7, 10, 11</sup></span> Notably, trisomy 8q was accompanied by inv(14)(q11q32) in four of our cases. Also, four of our five patients (case 1, 2, 3, 5) exhibited 11q abnormalities, which are closely associated with <i>ATM</i> gene deletion. Our current findings support previous evidence indicating a T-PLL-specific synergistic relationship between <i>ATM</i> deletion and TCL1 overexpression. This may disrupt genomic stability, triggering leukocytosis and p53 activation.<span><sup>12</sup></span> Our findings contribute to a deeper understanding of the genetic and molecular mechanisms underlying T-PLL and we offer insights into factors that may influence disease progression and presentation.</p><p>However, our work had several limitations. First, the study was retrospective in nature. Second, we lacked karyotypic data on age-matched healthy controls as it is unethical to collect BM from healthy subjects. Third, three T-PLL patients with abnormalities involving chromosomes 7 and 14 did not undergo FISH testing. Fourth, only five T-PLL patients were included, more work is required.</p><p>In conclusion, the use of PHA-stimulated cultures for chromosomal karyotyping may be a substantial advance in screening the abnormal karyotypes of mature T lymphocytes. Our study underscores the need to use PHA-stimulated BM cell cultures in clinical settings for chromosomal karyotyping of patients with suspected abnormalities in their mature T lymphocyte clonalities. When diagnostic abnormalities are detected, further confirmatory tests can be performed.</p><p>F.G and S.C wrote the manuscript. F.G, C.M and Z.Y participated in the conception of the study. F.G, S.C, and J.L were instrumental in performing laboratory analysis, interpretation and discussion of data. C.M and Z.Y contributed to the clinical decision making. All authors reviewed and approved the final manuscript.</p><p>The authors declare no conflicts of interest.</p>","PeriodicalId":14120,"journal":{"name":"International Journal of Laboratory Hematology","volume":"46 5","pages":"975-977"},"PeriodicalIF":2.3000,"publicationDate":"2024-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/ijlh.14323","citationCount":"0","resultStr":"{\"title\":\"The critical role of phytohemagglutinin-stimulated cell cultures in the diagnosis of T-cell prolymphocytic leukemia: A case-based approach\",\"authors\":\"Fei Gao, Shuang Chen, Jianwei Li, Zailin Yang, Cui Mao\",\"doi\":\"10.1111/ijlh.14323\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>T-cell prolymphocytic leukemia (T-PLL) is a very rare subtype of the mature lymphocytic malignancies that typically occur in middle-aged and older individuals, accounting for approximately 2% of all mature T-cell lymphomas.<span><sup>1</sup></span> T-PLL is characterized by a poor median survival rate and distinctive cell morphological, immunophenotypic, and cytogenetic features. The major diagnostic criteria of the guidelines for T-PLL diagnosis<span><sup>2</sup></span> include: (1) 5 × 10<sup>9</sup>/L cells of the T-PLL phenotype in peripheral blood or bone marrow (BM); (2) T-cell clonality determined via PCR or flow cytometry (FCM); and, (3) an abnormal 14q32 or Xq28 karyotype or expression of <i>TCL1A/B</i> or <i>MTCP1</i>. Additionally, four minor criteria also contribute to the diagnostic framework: (1) abnormalities involving chromosome 11 (11q22.3; <i>ATM</i>); (2) abnormalities involving chromosome 8 such as idic(8)(p11), t(8;8), trisomy 8q; (3) abnormalities in chromosomes 5, 12, 13, 22, or a complex karyotype; and, (4) T-PLL-specific features (e.g., splenomegaly, effusions). A diagnosis of T-PLL is established if all three major criteria are met or if the first two such criteria and at least one minor criterion are met. Obviously, analysis of chromosomal abnormalities is essential for diagnosis of T-PLL. However, T-PLL poses a challenge in this regard given the low proliferation capacity of mature T lymphocytes, leading to a high failure rate of chromosomal karyotyping associated with conventional culture methods (short-term culture).</p><p>In this study, we retrospectively analyze and present five cases for whom we used a phytohemagglutinin (PHA)-stimulated culture method to increase the detection rate of chromosomal abnormalities in BM cells. This enhanced T-PLL diagnostic accuracy.</p><p>All five patients were males of median age 58 years (range, 42 to 83 years). Immunophenotypic abnormalities were observed in 50% ~ 90% of T cells of all patients (Table S1) in BM sample. Most exhibited the phenotype CD3<sup>+</sup>, CD2<sup>+</sup>, CD4<sup>+</sup>, CD5<sup>+</sup>, CD7<sup>+</sup>, CD56<sup>−</sup>, CD34<sup>−</sup>, CD117<sup>−</sup>, CD1a<sup>−</sup>, and CD52<sup>+</sup>. The most notable immunophenotypic difference was that for CD8: cases 1, 4, and 5 were of CD8<sup>part+</sup> status; case 2 CD8<sup>−</sup>; and case 3 CD8<sup>+</sup>. Subsequently, chromosomal karyotyping was performed on cultures grown with and without PHA (Table 1). Three of the five patients (cases 1, 2, and 3) exhibited only a 46,XY karyotype when cells were grown without PHA, but complex karyotypes, inv(14), trisomy 8q, and abnormalities in 11q when cells were grown with PHA. One patient (case 5) exhibited no metaphase cells when cells were grown without PHA, but a complex karyotype, abnormalities in 11q, and t (14;14) when cells were grown with PHA. Only one patient (case 4) yielded consistent chromosomal karyotypes when cells were grown with and without PHA, thus add(3)(q26), i(8)(q10), inv(14)(q11q32), add(16)(q22), and add(21)(p11). Figure 1 shows the chromosomal karyotypes afforded by the PHA-stimulated cell culture approach for all cases. Obviously, the use of PHA-stimulated BM cell cultures for chromosomal karyotyping significantly increased the detection rates of chromosomal abnormalities (4/5, 80%). PHA, a pan-lymphocyte mitogen, effectively stimulates T cell proliferation and differentiation. Although the properties of PHA-conditioned medium have been described in previous studies,<span><sup>3-5</sup></span> culture of BM cells with PHA is not commonly used in clinical practice for chromosomal karyotyping of patients under suspicion of abnormal, mature T lymphocyte status. Our results further emphasize that PHA-stimulated cell culture enhances the detection of chromosomal abnormalities in T-PLL patients.</p><p>Strikingly, we found that four of the five patients (cases 1, 2, 3, and 4) were of inv(14)(q11q32) status and one (case 5) of t(14;14)(q11;q32) status. Dürig et al. reported that 80% of T-PLL patients exhibited complex chromosomal karyotypes such as inv(14)(q11q32).<span><sup>6</sup></span> The 14q32 abnormality frequently triggers formation of the <i>TRA-TRD::TCL1A</i> fusion gene, a notable genetic aberration of T-PLL.<span><sup>7-9</sup></span> In two of our patients (cases 1, 4), <i>TCL1</i> rearrangements were identified via fluorescence in situ hybridization (FISH) (Figure S1). Furthermore, abnormalities in chromosomes 8 and 12p, and deletions in the long arms of chromosomes 5, 6, 11, and 13, have been reported in T-PLL patients.<span><sup>7</sup></span> Several studies also suggested that 40% ~ 80% of T-PLL patients exhibit trisomy 8q.<span><sup>7, 10, 11</sup></span> Notably, trisomy 8q was accompanied by inv(14)(q11q32) in four of our cases. Also, four of our five patients (case 1, 2, 3, 5) exhibited 11q abnormalities, which are closely associated with <i>ATM</i> gene deletion. Our current findings support previous evidence indicating a T-PLL-specific synergistic relationship between <i>ATM</i> deletion and TCL1 overexpression. This may disrupt genomic stability, triggering leukocytosis and p53 activation.<span><sup>12</sup></span> Our findings contribute to a deeper understanding of the genetic and molecular mechanisms underlying T-PLL and we offer insights into factors that may influence disease progression and presentation.</p><p>However, our work had several limitations. First, the study was retrospective in nature. Second, we lacked karyotypic data on age-matched healthy controls as it is unethical to collect BM from healthy subjects. Third, three T-PLL patients with abnormalities involving chromosomes 7 and 14 did not undergo FISH testing. Fourth, only five T-PLL patients were included, more work is required.</p><p>In conclusion, the use of PHA-stimulated cultures for chromosomal karyotyping may be a substantial advance in screening the abnormal karyotypes of mature T lymphocytes. Our study underscores the need to use PHA-stimulated BM cell cultures in clinical settings for chromosomal karyotyping of patients with suspected abnormalities in their mature T lymphocyte clonalities. When diagnostic abnormalities are detected, further confirmatory tests can be performed.</p><p>F.G and S.C wrote the manuscript. F.G, C.M and Z.Y participated in the conception of the study. F.G, S.C, and J.L were instrumental in performing laboratory analysis, interpretation and discussion of data. C.M and Z.Y contributed to the clinical decision making. All authors reviewed and approved the final manuscript.</p><p>The authors declare no conflicts of interest.</p>\",\"PeriodicalId\":14120,\"journal\":{\"name\":\"International Journal of Laboratory Hematology\",\"volume\":\"46 5\",\"pages\":\"975-977\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2024-06-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/ijlh.14323\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Laboratory Hematology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/ijlh.14323\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Laboratory Hematology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/ijlh.14323","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
12 我们的研究结果有助于加深对 T-PLL 的遗传和分子机制的理解,我们还对可能影响疾病进展和表现的因素提出了见解。首先,这项研究属于回顾性研究。其次,我们缺乏年龄匹配的健康对照组的核型数据,因为从健康受试者身上收集骨髓是不道德的。第三,3 名涉及 7 号和 14 号染色体异常的 T-PLL 患者没有进行 FISH 检测。总之,使用 PHA 刺激培养物进行染色体核型分析可能是筛查成熟 T 淋巴细胞异常核型的一大进步。我们的研究强调了在临床环境中使用 PHA 刺激的 BM 细胞培养物对成熟 T 淋巴细胞克隆疑似异常的患者进行染色体核型分析的必要性。当检测到诊断异常时,可进行进一步的确证试验。F.G、C.M和Z.Y参与了研究的构思。F.G、S.C和J.L参与了实验分析、数据解释和讨论。C.M 和 Z.Y 参与了临床决策。所有作者均审阅并批准了最终手稿。
The critical role of phytohemagglutinin-stimulated cell cultures in the diagnosis of T-cell prolymphocytic leukemia: A case-based approach
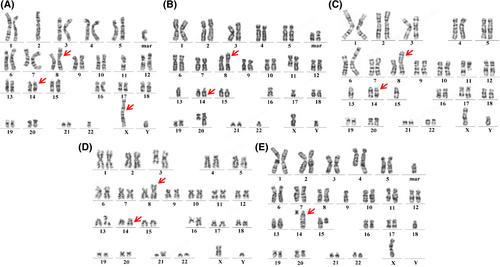
T-cell prolymphocytic leukemia (T-PLL) is a very rare subtype of the mature lymphocytic malignancies that typically occur in middle-aged and older individuals, accounting for approximately 2% of all mature T-cell lymphomas.1 T-PLL is characterized by a poor median survival rate and distinctive cell morphological, immunophenotypic, and cytogenetic features. The major diagnostic criteria of the guidelines for T-PLL diagnosis2 include: (1) 5 × 109/L cells of the T-PLL phenotype in peripheral blood or bone marrow (BM); (2) T-cell clonality determined via PCR or flow cytometry (FCM); and, (3) an abnormal 14q32 or Xq28 karyotype or expression of TCL1A/B or MTCP1. Additionally, four minor criteria also contribute to the diagnostic framework: (1) abnormalities involving chromosome 11 (11q22.3; ATM); (2) abnormalities involving chromosome 8 such as idic(8)(p11), t(8;8), trisomy 8q; (3) abnormalities in chromosomes 5, 12, 13, 22, or a complex karyotype; and, (4) T-PLL-specific features (e.g., splenomegaly, effusions). A diagnosis of T-PLL is established if all three major criteria are met or if the first two such criteria and at least one minor criterion are met. Obviously, analysis of chromosomal abnormalities is essential for diagnosis of T-PLL. However, T-PLL poses a challenge in this regard given the low proliferation capacity of mature T lymphocytes, leading to a high failure rate of chromosomal karyotyping associated with conventional culture methods (short-term culture).
In this study, we retrospectively analyze and present five cases for whom we used a phytohemagglutinin (PHA)-stimulated culture method to increase the detection rate of chromosomal abnormalities in BM cells. This enhanced T-PLL diagnostic accuracy.
All five patients were males of median age 58 years (range, 42 to 83 years). Immunophenotypic abnormalities were observed in 50% ~ 90% of T cells of all patients (Table S1) in BM sample. Most exhibited the phenotype CD3+, CD2+, CD4+, CD5+, CD7+, CD56−, CD34−, CD117−, CD1a−, and CD52+. The most notable immunophenotypic difference was that for CD8: cases 1, 4, and 5 were of CD8part+ status; case 2 CD8−; and case 3 CD8+. Subsequently, chromosomal karyotyping was performed on cultures grown with and without PHA (Table 1). Three of the five patients (cases 1, 2, and 3) exhibited only a 46,XY karyotype when cells were grown without PHA, but complex karyotypes, inv(14), trisomy 8q, and abnormalities in 11q when cells were grown with PHA. One patient (case 5) exhibited no metaphase cells when cells were grown without PHA, but a complex karyotype, abnormalities in 11q, and t (14;14) when cells were grown with PHA. Only one patient (case 4) yielded consistent chromosomal karyotypes when cells were grown with and without PHA, thus add(3)(q26), i(8)(q10), inv(14)(q11q32), add(16)(q22), and add(21)(p11). Figure 1 shows the chromosomal karyotypes afforded by the PHA-stimulated cell culture approach for all cases. Obviously, the use of PHA-stimulated BM cell cultures for chromosomal karyotyping significantly increased the detection rates of chromosomal abnormalities (4/5, 80%). PHA, a pan-lymphocyte mitogen, effectively stimulates T cell proliferation and differentiation. Although the properties of PHA-conditioned medium have been described in previous studies,3-5 culture of BM cells with PHA is not commonly used in clinical practice for chromosomal karyotyping of patients under suspicion of abnormal, mature T lymphocyte status. Our results further emphasize that PHA-stimulated cell culture enhances the detection of chromosomal abnormalities in T-PLL patients.
Strikingly, we found that four of the five patients (cases 1, 2, 3, and 4) were of inv(14)(q11q32) status and one (case 5) of t(14;14)(q11;q32) status. Dürig et al. reported that 80% of T-PLL patients exhibited complex chromosomal karyotypes such as inv(14)(q11q32).6 The 14q32 abnormality frequently triggers formation of the TRA-TRD::TCL1A fusion gene, a notable genetic aberration of T-PLL.7-9 In two of our patients (cases 1, 4), TCL1 rearrangements were identified via fluorescence in situ hybridization (FISH) (Figure S1). Furthermore, abnormalities in chromosomes 8 and 12p, and deletions in the long arms of chromosomes 5, 6, 11, and 13, have been reported in T-PLL patients.7 Several studies also suggested that 40% ~ 80% of T-PLL patients exhibit trisomy 8q.7, 10, 11 Notably, trisomy 8q was accompanied by inv(14)(q11q32) in four of our cases. Also, four of our five patients (case 1, 2, 3, 5) exhibited 11q abnormalities, which are closely associated with ATM gene deletion. Our current findings support previous evidence indicating a T-PLL-specific synergistic relationship between ATM deletion and TCL1 overexpression. This may disrupt genomic stability, triggering leukocytosis and p53 activation.12 Our findings contribute to a deeper understanding of the genetic and molecular mechanisms underlying T-PLL and we offer insights into factors that may influence disease progression and presentation.
However, our work had several limitations. First, the study was retrospective in nature. Second, we lacked karyotypic data on age-matched healthy controls as it is unethical to collect BM from healthy subjects. Third, three T-PLL patients with abnormalities involving chromosomes 7 and 14 did not undergo FISH testing. Fourth, only five T-PLL patients were included, more work is required.
In conclusion, the use of PHA-stimulated cultures for chromosomal karyotyping may be a substantial advance in screening the abnormal karyotypes of mature T lymphocytes. Our study underscores the need to use PHA-stimulated BM cell cultures in clinical settings for chromosomal karyotyping of patients with suspected abnormalities in their mature T lymphocyte clonalities. When diagnostic abnormalities are detected, further confirmatory tests can be performed.
F.G and S.C wrote the manuscript. F.G, C.M and Z.Y participated in the conception of the study. F.G, S.C, and J.L were instrumental in performing laboratory analysis, interpretation and discussion of data. C.M and Z.Y contributed to the clinical decision making. All authors reviewed and approved the final manuscript.
期刊介绍:
The International Journal of Laboratory Hematology provides a forum for the communication of new developments, research topics and the practice of laboratory haematology.
The journal publishes invited reviews, full length original articles, and correspondence.
The International Journal of Laboratory Hematology is the official journal of the International Society for Laboratory Hematology, which addresses the following sub-disciplines: cellular analysis, flow cytometry, haemostasis and thrombosis, molecular diagnostics, haematology informatics, haemoglobinopathies, point of care testing, standards and guidelines.
The journal was launched in 2006 as the successor to Clinical and Laboratory Hematology, which was first published in 1979. An active and positive editorial policy ensures that work of a high scientific standard is reported, in order to bridge the gap between practical and academic aspects of laboratory haematology.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
