{"title":"TS-tools:根据文本反应 SMILES 输入快速自动定位过渡状态。","authors":"Thijs Stuyver","doi":"10.1002/jcc.27374","DOIUrl":null,"url":null,"abstract":"<p>Here, TS-tools is presented, a Python package facilitating the automated localization of transition states (TS) based on a textual reaction SMILES input. TS searches can either be performed at xTB or DFT level of theory, with the former yielding guesses at marginal computational cost, and the latter directly yielding accurate structures at greater expense. On a benchmarking dataset of mono- and bimolecular reactions, TS-tools reaches an excellent success rate of 95% already at xTB level of theory. For tri- and multimolecular reaction pathways - which are typically not benchmarked when developing new automated TS search approaches, yet are relevant for various types of reactivity, cf. solvent- and autocatalysis and enzymatic reactivity - TS-tools retains its ability to identify TS geometries, though a DFT treatment becomes essential in many cases. Throughout the presented applications, a particular emphasis is placed on solvation-induced mechanistic changes, another issue that received limited attention in the automated TS search literature so far.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 27","pages":"2308-2317"},"PeriodicalIF":3.4000,"publicationDate":"2024-06-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27374","citationCount":"0","resultStr":"{\"title\":\"TS-tools: Rapid and automated localization of transition states based on a textual reaction SMILES input\",\"authors\":\"Thijs Stuyver\",\"doi\":\"10.1002/jcc.27374\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Here, TS-tools is presented, a Python package facilitating the automated localization of transition states (TS) based on a textual reaction SMILES input. TS searches can either be performed at xTB or DFT level of theory, with the former yielding guesses at marginal computational cost, and the latter directly yielding accurate structures at greater expense. On a benchmarking dataset of mono- and bimolecular reactions, TS-tools reaches an excellent success rate of 95% already at xTB level of theory. For tri- and multimolecular reaction pathways - which are typically not benchmarked when developing new automated TS search approaches, yet are relevant for various types of reactivity, cf. solvent- and autocatalysis and enzymatic reactivity - TS-tools retains its ability to identify TS geometries, though a DFT treatment becomes essential in many cases. Throughout the presented applications, a particular emphasis is placed on solvation-induced mechanistic changes, another issue that received limited attention in the automated TS search literature so far.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 27\",\"pages\":\"2308-2317\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-06-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27374\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27374\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27374","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
TS-tools: Rapid and automated localization of transition states based on a textual reaction SMILES input
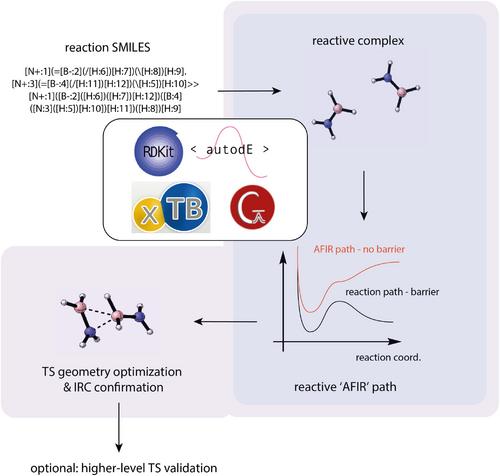
Here, TS-tools is presented, a Python package facilitating the automated localization of transition states (TS) based on a textual reaction SMILES input. TS searches can either be performed at xTB or DFT level of theory, with the former yielding guesses at marginal computational cost, and the latter directly yielding accurate structures at greater expense. On a benchmarking dataset of mono- and bimolecular reactions, TS-tools reaches an excellent success rate of 95% already at xTB level of theory. For tri- and multimolecular reaction pathways - which are typically not benchmarked when developing new automated TS search approaches, yet are relevant for various types of reactivity, cf. solvent- and autocatalysis and enzymatic reactivity - TS-tools retains its ability to identify TS geometries, though a DFT treatment becomes essential in many cases. Throughout the presented applications, a particular emphasis is placed on solvation-induced mechanistic changes, another issue that received limited attention in the automated TS search literature so far.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
