{"title":"作为 Selinexor 药物跨细胞膜输送系统的工程纳米粒子以及癌细胞中的相关信号通路","authors":"Alireza Nakhaei, Heidar Raissi, Farzaneh Farzad","doi":"10.1016/j.jmgm.2024.108809","DOIUrl":null,"url":null,"abstract":"<div><p>In the present work, molecular dynamics simulation is applied to evaluate the drug carrier efficiency of graphene oxide nanoflake (GONF) for loading of Selinexor (SXR) drug as well as the drug delivery by 2D material through the membrane in aqueous solution. In addition, to investigate the adsorption and penetration of drug-nanocarrier complex into the cell membrane, well-tempered metadynamics simulations and steered molecular dynamics (SMD) simulations were performed. Based on the obtained results, it is evident that intermolecular hydrogen bonds (HBs) and π-π interactions play a significant role in expediting the interaction between drug molecules and the graphene oxide (GO) nanosheet, ultimately resulting in the formation of a stable SXR-GO complex. The Lennard-Jones (L-J) energy value for the interaction of SXR with GONF is calculated to be approximately −98.85 kJ/mol. In the SXR-GONF complex system, the dominant interaction between SXR and GONF is attributed to the L-J term, resulting from the formation of a strong π−π interaction between the drug molecules and the substrate surface. Moreover, our simulations show by decreasing the distance of GONF with respect to cell membrane, the interaction energy of GONF-membrane significantly decrease to −1500 kJ/mol resulting in fast diffusion of SXR-GONF complex toward the bilayer surface that is favored opening the way to natural drug nanocapsule.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"131 ","pages":"Article 108809"},"PeriodicalIF":3.0000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Engineered nanoparticles as Selinexor drug delivery systems across the cell membrane and related signaling pathways in cancer cells\",\"authors\":\"Alireza Nakhaei, Heidar Raissi, Farzaneh Farzad\",\"doi\":\"10.1016/j.jmgm.2024.108809\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>In the present work, molecular dynamics simulation is applied to evaluate the drug carrier efficiency of graphene oxide nanoflake (GONF) for loading of Selinexor (SXR) drug as well as the drug delivery by 2D material through the membrane in aqueous solution. In addition, to investigate the adsorption and penetration of drug-nanocarrier complex into the cell membrane, well-tempered metadynamics simulations and steered molecular dynamics (SMD) simulations were performed. Based on the obtained results, it is evident that intermolecular hydrogen bonds (HBs) and π-π interactions play a significant role in expediting the interaction between drug molecules and the graphene oxide (GO) nanosheet, ultimately resulting in the formation of a stable SXR-GO complex. The Lennard-Jones (L-J) energy value for the interaction of SXR with GONF is calculated to be approximately −98.85 kJ/mol. In the SXR-GONF complex system, the dominant interaction between SXR and GONF is attributed to the L-J term, resulting from the formation of a strong π−π interaction between the drug molecules and the substrate surface. Moreover, our simulations show by decreasing the distance of GONF with respect to cell membrane, the interaction energy of GONF-membrane significantly decrease to −1500 kJ/mol resulting in fast diffusion of SXR-GONF complex toward the bilayer surface that is favored opening the way to natural drug nanocapsule.</p></div>\",\"PeriodicalId\":16361,\"journal\":{\"name\":\"Journal of molecular graphics & modelling\",\"volume\":\"131 \",\"pages\":\"Article 108809\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular graphics & modelling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1093326324001098\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/6/13 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001098","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/13 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Engineered nanoparticles as Selinexor drug delivery systems across the cell membrane and related signaling pathways in cancer cells
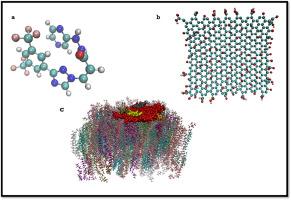
In the present work, molecular dynamics simulation is applied to evaluate the drug carrier efficiency of graphene oxide nanoflake (GONF) for loading of Selinexor (SXR) drug as well as the drug delivery by 2D material through the membrane in aqueous solution. In addition, to investigate the adsorption and penetration of drug-nanocarrier complex into the cell membrane, well-tempered metadynamics simulations and steered molecular dynamics (SMD) simulations were performed. Based on the obtained results, it is evident that intermolecular hydrogen bonds (HBs) and π-π interactions play a significant role in expediting the interaction between drug molecules and the graphene oxide (GO) nanosheet, ultimately resulting in the formation of a stable SXR-GO complex. The Lennard-Jones (L-J) energy value for the interaction of SXR with GONF is calculated to be approximately −98.85 kJ/mol. In the SXR-GONF complex system, the dominant interaction between SXR and GONF is attributed to the L-J term, resulting from the formation of a strong π−π interaction between the drug molecules and the substrate surface. Moreover, our simulations show by decreasing the distance of GONF with respect to cell membrane, the interaction energy of GONF-membrane significantly decrease to −1500 kJ/mol resulting in fast diffusion of SXR-GONF complex toward the bilayer surface that is favored opening the way to natural drug nanocapsule.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
