Jessica L. Taylor, Pedro H. Ayres-Galhardo and Breann L. Brown*,
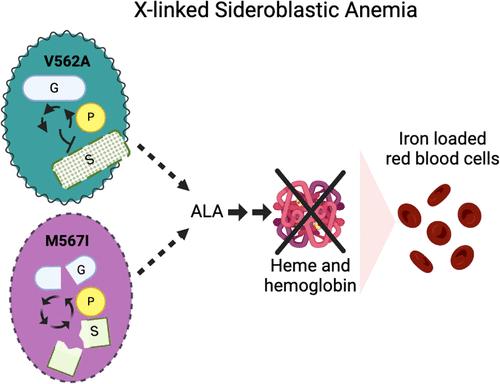
{"title":"阐明人类 ALAS2 C 端突变导致功能丧失和疾病的作用","authors":"Jessica L. Taylor, Pedro H. Ayres-Galhardo and Breann L. Brown*, ","doi":"10.1021/acs.biochem.4c00066","DOIUrl":null,"url":null,"abstract":"<p >The conserved enzyme aminolevulinic acid synthase (ALAS) initiates heme biosynthesis in certain bacteria and eukaryotes by catalyzing the condensation of glycine and succinyl-CoA to yield aminolevulinic acid. In humans, the ALAS isoform responsible for heme production during red blood cell development is the erythroid-specific ALAS2 isoform. Owing to its essential role in erythropoiesis, changes in human ALAS2 (hALAS2) function can lead to two different blood disorders. X-linked sideroblastic anemia results from loss of ALAS2 function, while X-linked protoporphyria results from gain of ALAS2 function. Interestingly, mutations in the ALAS2 C-terminal extension can be implicated in both diseases. Here, we investigate the molecular basis for enzyme dysfunction mediated by two previously reported C-terminal loss-of-function variants, hALAS2 V562A and M567I. We show that the mutations do not result in gross structural perturbations, but the enzyme stability for V562A is decreased. Additionally, we show that enzyme stability moderately increases with the addition of the pyridoxal 5′-phosphate (PLP) cofactor for both variants. The variants display differential binding to PLP and the individual substrates compared to wild-type hALAS2. Although hALAS2 V562A is a more active enzyme <i>in vitro</i>, it is less efficient concerning succinyl-CoA binding. In contrast, the M567I mutation significantly alters the cooperativity of substrate binding. In combination with previously reported cell-based studies, our work reveals the molecular basis by which hALAS2 C-terminal mutations negatively affect ALA production necessary for proper heme biosynthesis.</p>","PeriodicalId":28,"journal":{"name":"Biochemistry Biochemistry","volume":"63 13","pages":"1636–1646"},"PeriodicalIF":2.7000,"publicationDate":"2024-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.biochem.4c00066","citationCount":"0","resultStr":"{\"title\":\"Elucidating the Role of Human ALAS2 C-terminal Mutations Resulting in Loss of Function and Disease\",\"authors\":\"Jessica L. Taylor, Pedro H. Ayres-Galhardo and Breann L. Brown*, \",\"doi\":\"10.1021/acs.biochem.4c00066\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The conserved enzyme aminolevulinic acid synthase (ALAS) initiates heme biosynthesis in certain bacteria and eukaryotes by catalyzing the condensation of glycine and succinyl-CoA to yield aminolevulinic acid. In humans, the ALAS isoform responsible for heme production during red blood cell development is the erythroid-specific ALAS2 isoform. Owing to its essential role in erythropoiesis, changes in human ALAS2 (hALAS2) function can lead to two different blood disorders. X-linked sideroblastic anemia results from loss of ALAS2 function, while X-linked protoporphyria results from gain of ALAS2 function. Interestingly, mutations in the ALAS2 C-terminal extension can be implicated in both diseases. Here, we investigate the molecular basis for enzyme dysfunction mediated by two previously reported C-terminal loss-of-function variants, hALAS2 V562A and M567I. We show that the mutations do not result in gross structural perturbations, but the enzyme stability for V562A is decreased. Additionally, we show that enzyme stability moderately increases with the addition of the pyridoxal 5′-phosphate (PLP) cofactor for both variants. The variants display differential binding to PLP and the individual substrates compared to wild-type hALAS2. Although hALAS2 V562A is a more active enzyme <i>in vitro</i>, it is less efficient concerning succinyl-CoA binding. In contrast, the M567I mutation significantly alters the cooperativity of substrate binding. In combination with previously reported cell-based studies, our work reveals the molecular basis by which hALAS2 C-terminal mutations negatively affect ALA production necessary for proper heme biosynthesis.</p>\",\"PeriodicalId\":28,\"journal\":{\"name\":\"Biochemistry Biochemistry\",\"volume\":\"63 13\",\"pages\":\"1636–1646\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-06-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.biochem.4c00066\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biochemistry Biochemistry\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.biochem.4c00066\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biochemistry Biochemistry","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.biochem.4c00066","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
在某些细菌和真核生物中,氨基乙酰丙酸合成酶(ALAS)通过催化甘氨酸和琥珀酰-CoA缩合生成氨基乙酰丙酸,从而启动血红素的生物合成。在人类,负责在红细胞发育过程中产生血红素的 ALAS 同工酶是红细胞特异性 ALAS2 同工酶。由于 ALAS2 在红细胞生成过程中的重要作用,人类 ALAS2(hALAS2)功能的变化可导致两种不同的血液疾病。X连锁性红细胞性贫血是由ALAS2功能缺失引起的,而X连锁性原卟啉症则是由ALAS2功能获得引起的。有趣的是,ALAS2 C端延伸部分的突变可能与这两种疾病有关。在这里,我们研究了之前报道的两种 C 端功能缺失变体(hALAS2 V562A 和 M567I)介导的酶功能障碍的分子基础。我们发现,这些变异不会导致严重的结构紊乱,但 V562A 的酶稳定性会降低。此外,我们还发现这两个变体在加入 5'-磷酸吡哆醛(PLP)辅助因子后,酶的稳定性会适度增加。与野生型 hALAS2 相比,这些变体与 PLP 和单个底物的结合存在差异。虽然 hALAS2 V562A 在体外是一种更活跃的酶,但它与琥珀酰-CoA 结合的效率较低。相比之下,M567I 突变显著改变了底物结合的合作性。结合之前报道的基于细胞的研究,我们的工作揭示了 hALAS2 C 端突变对正常血红素生物合成所需的 ALA 生成产生负面影响的分子基础。
Elucidating the Role of Human ALAS2 C-terminal Mutations Resulting in Loss of Function and Disease
The conserved enzyme aminolevulinic acid synthase (ALAS) initiates heme biosynthesis in certain bacteria and eukaryotes by catalyzing the condensation of glycine and succinyl-CoA to yield aminolevulinic acid. In humans, the ALAS isoform responsible for heme production during red blood cell development is the erythroid-specific ALAS2 isoform. Owing to its essential role in erythropoiesis, changes in human ALAS2 (hALAS2) function can lead to two different blood disorders. X-linked sideroblastic anemia results from loss of ALAS2 function, while X-linked protoporphyria results from gain of ALAS2 function. Interestingly, mutations in the ALAS2 C-terminal extension can be implicated in both diseases. Here, we investigate the molecular basis for enzyme dysfunction mediated by two previously reported C-terminal loss-of-function variants, hALAS2 V562A and M567I. We show that the mutations do not result in gross structural perturbations, but the enzyme stability for V562A is decreased. Additionally, we show that enzyme stability moderately increases with the addition of the pyridoxal 5′-phosphate (PLP) cofactor for both variants. The variants display differential binding to PLP and the individual substrates compared to wild-type hALAS2. Although hALAS2 V562A is a more active enzyme in vitro, it is less efficient concerning succinyl-CoA binding. In contrast, the M567I mutation significantly alters the cooperativity of substrate binding. In combination with previously reported cell-based studies, our work reveals the molecular basis by which hALAS2 C-terminal mutations negatively affect ALA production necessary for proper heme biosynthesis.
期刊介绍:
Biochemistry provides an international forum for publishing exceptional, rigorous, high-impact research across all of biological chemistry. This broad scope includes studies on the chemical, physical, mechanistic, and/or structural basis of biological or cell function, and encompasses the fields of chemical biology, synthetic biology, disease biology, cell biology, nucleic acid biology, neuroscience, structural biology, and biophysics. In addition to traditional Research Articles, Biochemistry also publishes Communications, Viewpoints, and Perspectives, as well as From the Bench articles that report new methods of particular interest to the biological chemistry community.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
