Yuanyuan Zhang, Haiming Gao, Lu Zhang, Yunjing Zhao, Chuang Qiu, Xiaoliang Liu
{"title":"重度胼胝体畸形家族中的新型基因 KIT 变异:病例系列和文献综述","authors":"Yuanyuan Zhang, Haiming Gao, Lu Zhang, Yunjing Zhao, Chuang Qiu, Xiaoliang Liu","doi":"10.1002/jcla.25073","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Introduction</h3>\n \n <p>Piebaldism is a rare autosomal dominant disorder characterized by congenital white forelock and depigmented patches, which is most commonly caused by deleterious variants in the <i>KIT</i> gene.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>Four <i>KIT</i> variants were identified in a piebaldism case series by whole-exome sequencing. Functional experiments, including in vitro minigene reporter assay and enzyme-linked immunosorbent assay, were carried out to elucidate the pathogenicity of the variants. The genotype–phenotype correlation was summarized through extensive literature reviewing.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>All the four cases had severe piebaldism presented with typical white forelock and diffuse depigmentation on the ventral trunk and limbs. Four germline variants at the tyrosine kinase (TK) domains of the <i>KIT</i> gene were identified: two novel variants c.1990+1G>A (p.Pro627_Gly664delinsArg) and c.2716T>C (p.Cys906Arg), and two known variants c.1879+1G>A (p.Gly592_Pro627delinsAla) and c.1747G>A (p.Glu583Lys). Both splicing variants caused exon skipping and inframe deletions in the TK1 domain. The missense variants resided at the TK1 and TK2 domains respectively impairing PI3K/AKT and MAPK/ERK signaling pathways, the downstream of KIT. All severe cases were associated with variants in the TK domains, eliciting a major dominant-negative mechanism of the disease.</p>\n </section>\n \n <section>\n \n <h3> Conclusion</h3>\n \n <p>Our data expand the mutation spectrum of <i>KIT</i>, emphasized by a dominant-negative effect of variants in the critical TK domains in severe cases. We also share the experience of prenatal diagnosis and informed reproductive choices for the affected families.</p>\n </section>\n </div>","PeriodicalId":15509,"journal":{"name":"Journal of Clinical Laboratory Analysis","volume":"38 11-12","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-06-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcla.25073","citationCount":"0","resultStr":"{\"title\":\"Novel Germline KIT Variants in Families With Severe Piebaldism: Case Series and Literature Review\",\"authors\":\"Yuanyuan Zhang, Haiming Gao, Lu Zhang, Yunjing Zhao, Chuang Qiu, Xiaoliang Liu\",\"doi\":\"10.1002/jcla.25073\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Introduction</h3>\\n \\n <p>Piebaldism is a rare autosomal dominant disorder characterized by congenital white forelock and depigmented patches, which is most commonly caused by deleterious variants in the <i>KIT</i> gene.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>Four <i>KIT</i> variants were identified in a piebaldism case series by whole-exome sequencing. Functional experiments, including in vitro minigene reporter assay and enzyme-linked immunosorbent assay, were carried out to elucidate the pathogenicity of the variants. The genotype–phenotype correlation was summarized through extensive literature reviewing.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>All the four cases had severe piebaldism presented with typical white forelock and diffuse depigmentation on the ventral trunk and limbs. Four germline variants at the tyrosine kinase (TK) domains of the <i>KIT</i> gene were identified: two novel variants c.1990+1G>A (p.Pro627_Gly664delinsArg) and c.2716T>C (p.Cys906Arg), and two known variants c.1879+1G>A (p.Gly592_Pro627delinsAla) and c.1747G>A (p.Glu583Lys). Both splicing variants caused exon skipping and inframe deletions in the TK1 domain. The missense variants resided at the TK1 and TK2 domains respectively impairing PI3K/AKT and MAPK/ERK signaling pathways, the downstream of KIT. All severe cases were associated with variants in the TK domains, eliciting a major dominant-negative mechanism of the disease.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusion</h3>\\n \\n <p>Our data expand the mutation spectrum of <i>KIT</i>, emphasized by a dominant-negative effect of variants in the critical TK domains in severe cases. We also share the experience of prenatal diagnosis and informed reproductive choices for the affected families.</p>\\n </section>\\n </div>\",\"PeriodicalId\":15509,\"journal\":{\"name\":\"Journal of Clinical Laboratory Analysis\",\"volume\":\"38 11-12\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-06-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcla.25073\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Laboratory Analysis\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcla.25073\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MEDICAL LABORATORY TECHNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Laboratory Analysis","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcla.25073","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MEDICAL LABORATORY TECHNOLOGY","Score":null,"Total":0}
Novel Germline KIT Variants in Families With Severe Piebaldism: Case Series and Literature Review
Introduction
Piebaldism is a rare autosomal dominant disorder characterized by congenital white forelock and depigmented patches, which is most commonly caused by deleterious variants in the KIT gene.
Methods
Four KIT variants were identified in a piebaldism case series by whole-exome sequencing. Functional experiments, including in vitro minigene reporter assay and enzyme-linked immunosorbent assay, were carried out to elucidate the pathogenicity of the variants. The genotype–phenotype correlation was summarized through extensive literature reviewing.
Results
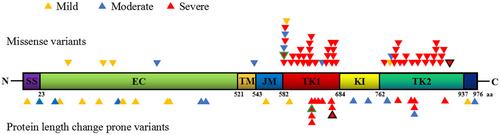
All the four cases had severe piebaldism presented with typical white forelock and diffuse depigmentation on the ventral trunk and limbs. Four germline variants at the tyrosine kinase (TK) domains of the KIT gene were identified: two novel variants c.1990+1G>A (p.Pro627_Gly664delinsArg) and c.2716T>C (p.Cys906Arg), and two known variants c.1879+1G>A (p.Gly592_Pro627delinsAla) and c.1747G>A (p.Glu583Lys). Both splicing variants caused exon skipping and inframe deletions in the TK1 domain. The missense variants resided at the TK1 and TK2 domains respectively impairing PI3K/AKT and MAPK/ERK signaling pathways, the downstream of KIT. All severe cases were associated with variants in the TK domains, eliciting a major dominant-negative mechanism of the disease.
Conclusion
Our data expand the mutation spectrum of KIT, emphasized by a dominant-negative effect of variants in the critical TK domains in severe cases. We also share the experience of prenatal diagnosis and informed reproductive choices for the affected families.
期刊介绍:
Journal of Clinical Laboratory Analysis publishes original articles on newly developing modes of technology and laboratory assays, with emphasis on their application in current and future clinical laboratory testing. This includes reports from the following fields: immunochemistry and toxicology, hematology and hematopathology, immunopathology, molecular diagnostics, microbiology, genetic testing, immunohematology, and clinical chemistry.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
