{"title":"有效种群数量如何说明等位基因变异的损失?","authors":"Fred W. Allendorf, Ola Hössjer, Nils Ryman","doi":"10.1111/eva.13733","DOIUrl":null,"url":null,"abstract":"<p>There are two primary measures of the amount of genetic variation in a population at a locus: heterozygosity and the number of alleles. Effective population size (<i>N</i><sub>e</sub>) provides both an expectation of the amount of heterozygosity in a population at drift-mutation equilibrium and the rate of loss of heterozygosity because of genetic drift. In contrast, the number of alleles in a population at drift-mutation equilibrium is a function of both <i>N</i><sub>e</sub> and census size (<i>N</i><sub>C</sub>). In addition, populations with the same <i>N</i><sub>e</sub> can lose allelic variation at very different rates. Allelic variation is generally much more sensitive to bottlenecks than heterozygosity. Expressions used to adjust for the effects of violations of the ideal population on <i>N</i><sub>e</sub> do not provide good predictions of the loss of allelic variation. These effects are much greater for loci with many alleles, which are often important for adaptation. We show that there is a linear relationship between the reduction of <i>N</i><sub>C</sub> and the corresponding reduction of the expected number of alleles at drift-mutation equilibrium. This makes it possible to predict the expected effect of a bottleneck on allelic variation. Heterozygosity provides good estimates of the rate of adaptive change in the short-term, but allelic variation provides important information about long-term adaptive change. The guideline of long-term <i>N</i><sub>e</sub> being greater than 500 is often used as a primary genetic metric for evaluating conservation status. We recommend that this guideline be expanded to take into account allelic variation as well as heterozygosity.</p>","PeriodicalId":168,"journal":{"name":"Evolutionary Applications","volume":"17 6","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-06-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/eva.13733","citationCount":"0","resultStr":"{\"title\":\"What does effective population size tell us about loss of allelic variation?\",\"authors\":\"Fred W. Allendorf, Ola Hössjer, Nils Ryman\",\"doi\":\"10.1111/eva.13733\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>There are two primary measures of the amount of genetic variation in a population at a locus: heterozygosity and the number of alleles. Effective population size (<i>N</i><sub>e</sub>) provides both an expectation of the amount of heterozygosity in a population at drift-mutation equilibrium and the rate of loss of heterozygosity because of genetic drift. In contrast, the number of alleles in a population at drift-mutation equilibrium is a function of both <i>N</i><sub>e</sub> and census size (<i>N</i><sub>C</sub>). In addition, populations with the same <i>N</i><sub>e</sub> can lose allelic variation at very different rates. Allelic variation is generally much more sensitive to bottlenecks than heterozygosity. Expressions used to adjust for the effects of violations of the ideal population on <i>N</i><sub>e</sub> do not provide good predictions of the loss of allelic variation. These effects are much greater for loci with many alleles, which are often important for adaptation. We show that there is a linear relationship between the reduction of <i>N</i><sub>C</sub> and the corresponding reduction of the expected number of alleles at drift-mutation equilibrium. This makes it possible to predict the expected effect of a bottleneck on allelic variation. Heterozygosity provides good estimates of the rate of adaptive change in the short-term, but allelic variation provides important information about long-term adaptive change. The guideline of long-term <i>N</i><sub>e</sub> being greater than 500 is often used as a primary genetic metric for evaluating conservation status. We recommend that this guideline be expanded to take into account allelic variation as well as heterozygosity.</p>\",\"PeriodicalId\":168,\"journal\":{\"name\":\"Evolutionary Applications\",\"volume\":\"17 6\",\"pages\":\"\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-06-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/eva.13733\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Evolutionary Applications\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/eva.13733\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"EVOLUTIONARY BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary Applications","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/eva.13733","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
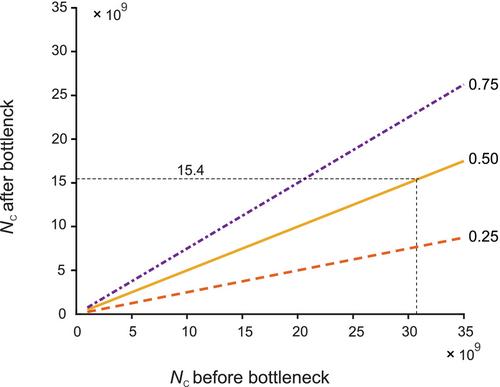
衡量一个位点上种群的遗传变异量有两个主要指标:杂合度和等位基因数。有效种群大小(Ne)提供了在漂变平衡状态下种群杂合度的期望值以及由于遗传漂变造成的杂合度损失率。相反,处于漂变平衡的种群中的等位基因数量是 Ne 和普查规模(NC)的函数。此外,具有相同 Ne 的种群的等位基因变异损失率也大不相同。等位基因变异通常比杂合度对瓶颈更为敏感。用来调整违反理想种群对 Ne 的影响的表达式并不能很好地预测等位基因变异的损失。这些影响对于等位基因较多的位点要大得多,而等位基因较多的位点通常对适应性很重要。我们的研究表明,在漂变平衡时,NC 的减少与等位基因预期数量的相应减少之间存在线性关系。这使得预测瓶颈对等位基因变异的预期影响成为可能。杂合度能很好地估计短期适应性变化的速度,但等位基因变异则能提供长期适应性变化的重要信息。长期 Ne 值大于 500 的准则通常被用作评估保护状况的主要遗传指标。我们建议扩大该准则的范围,将等位基因变异和杂合度也考虑在内。
What does effective population size tell us about loss of allelic variation?
There are two primary measures of the amount of genetic variation in a population at a locus: heterozygosity and the number of alleles. Effective population size (Ne) provides both an expectation of the amount of heterozygosity in a population at drift-mutation equilibrium and the rate of loss of heterozygosity because of genetic drift. In contrast, the number of alleles in a population at drift-mutation equilibrium is a function of both Ne and census size (NC). In addition, populations with the same Ne can lose allelic variation at very different rates. Allelic variation is generally much more sensitive to bottlenecks than heterozygosity. Expressions used to adjust for the effects of violations of the ideal population on Ne do not provide good predictions of the loss of allelic variation. These effects are much greater for loci with many alleles, which are often important for adaptation. We show that there is a linear relationship between the reduction of NC and the corresponding reduction of the expected number of alleles at drift-mutation equilibrium. This makes it possible to predict the expected effect of a bottleneck on allelic variation. Heterozygosity provides good estimates of the rate of adaptive change in the short-term, but allelic variation provides important information about long-term adaptive change. The guideline of long-term Ne being greater than 500 is often used as a primary genetic metric for evaluating conservation status. We recommend that this guideline be expanded to take into account allelic variation as well as heterozygosity.
期刊介绍:
Evolutionary Applications is a fully peer reviewed open access journal. It publishes papers that utilize concepts from evolutionary biology to address biological questions of health, social and economic relevance. Papers are expected to employ evolutionary concepts or methods to make contributions to areas such as (but not limited to): medicine, agriculture, forestry, exploitation and management (fisheries and wildlife), aquaculture, conservation biology, environmental sciences (including climate change and invasion biology), microbiology, and toxicology. All taxonomic groups are covered from microbes, fungi, plants and animals. In order to better serve the community, we also now strongly encourage submissions of papers making use of modern molecular and genetic methods (population and functional genomics, transcriptomics, proteomics, epigenetics, quantitative genetics, association and linkage mapping) to address important questions in any of these disciplines and in an applied evolutionary framework. Theoretical, empirical, synthesis or perspective papers are welcome.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
