Hugo Petitjean, Aude Giard, Jean-Pierre Flament, Catherine Berthomieu, Dorothée Berthomieu
{"title":"分子系统的快速振动分析","authors":"Hugo Petitjean, Aude Giard, Jean-Pierre Flament, Catherine Berthomieu, Dorothée Berthomieu","doi":"10.1002/jcc.27450","DOIUrl":null,"url":null,"abstract":"<p>The development of infrared difference spectroscopy provides unprecedented insights on structures of complex molecules like metalloproteins. However, the relevant information can be hard to find among the many bands of the vibrational spectra. The ab initio modeling is very helpful to assign the frequencies to vibrational modes but it is a challenge to process the huge quantity of data into descriptors useful for experimentalists. To this end, we developed a new tool called VIBMOL allowing to analyze vibrational modes of molecules from hessian matrices calculated with common quantum chemistry codes. VIBMOL program runs on Unix machines. Through a new graphical interface, the users can calculate the normal modes of molecules, visualize them, simulate infrared spectra, and explore the Potential Energy Distribution of normal modes among any set of vibration coordinates. It is combined with an interface program (gosdmu) formatting relevant data from the GAUSSIAN program. VIBMOL code is available upon request to the authors. A discussion is provided to help the readers to choose between a large choice of different software and it shows how VIBMOL can make the IR assignment easier in the context of collaborations with experimentalists.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 28","pages":"2374-2382"},"PeriodicalIF":2.9000,"publicationDate":"2024-06-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Fast vibrational analysis of molecular systems\",\"authors\":\"Hugo Petitjean, Aude Giard, Jean-Pierre Flament, Catherine Berthomieu, Dorothée Berthomieu\",\"doi\":\"10.1002/jcc.27450\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The development of infrared difference spectroscopy provides unprecedented insights on structures of complex molecules like metalloproteins. However, the relevant information can be hard to find among the many bands of the vibrational spectra. The ab initio modeling is very helpful to assign the frequencies to vibrational modes but it is a challenge to process the huge quantity of data into descriptors useful for experimentalists. To this end, we developed a new tool called VIBMOL allowing to analyze vibrational modes of molecules from hessian matrices calculated with common quantum chemistry codes. VIBMOL program runs on Unix machines. Through a new graphical interface, the users can calculate the normal modes of molecules, visualize them, simulate infrared spectra, and explore the Potential Energy Distribution of normal modes among any set of vibration coordinates. It is combined with an interface program (gosdmu) formatting relevant data from the GAUSSIAN program. VIBMOL code is available upon request to the authors. A discussion is provided to help the readers to choose between a large choice of different software and it shows how VIBMOL can make the IR assignment easier in the context of collaborations with experimentalists.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 28\",\"pages\":\"2374-2382\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-06-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27450\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27450","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
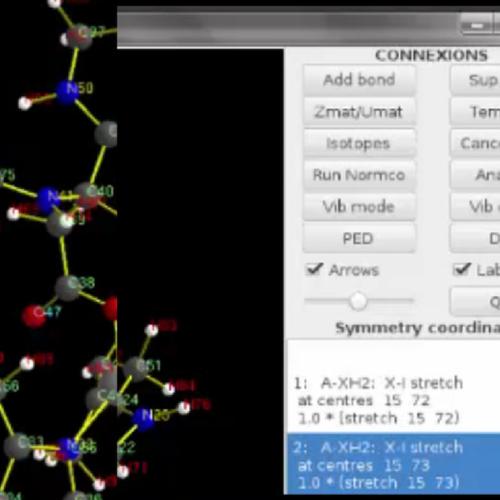
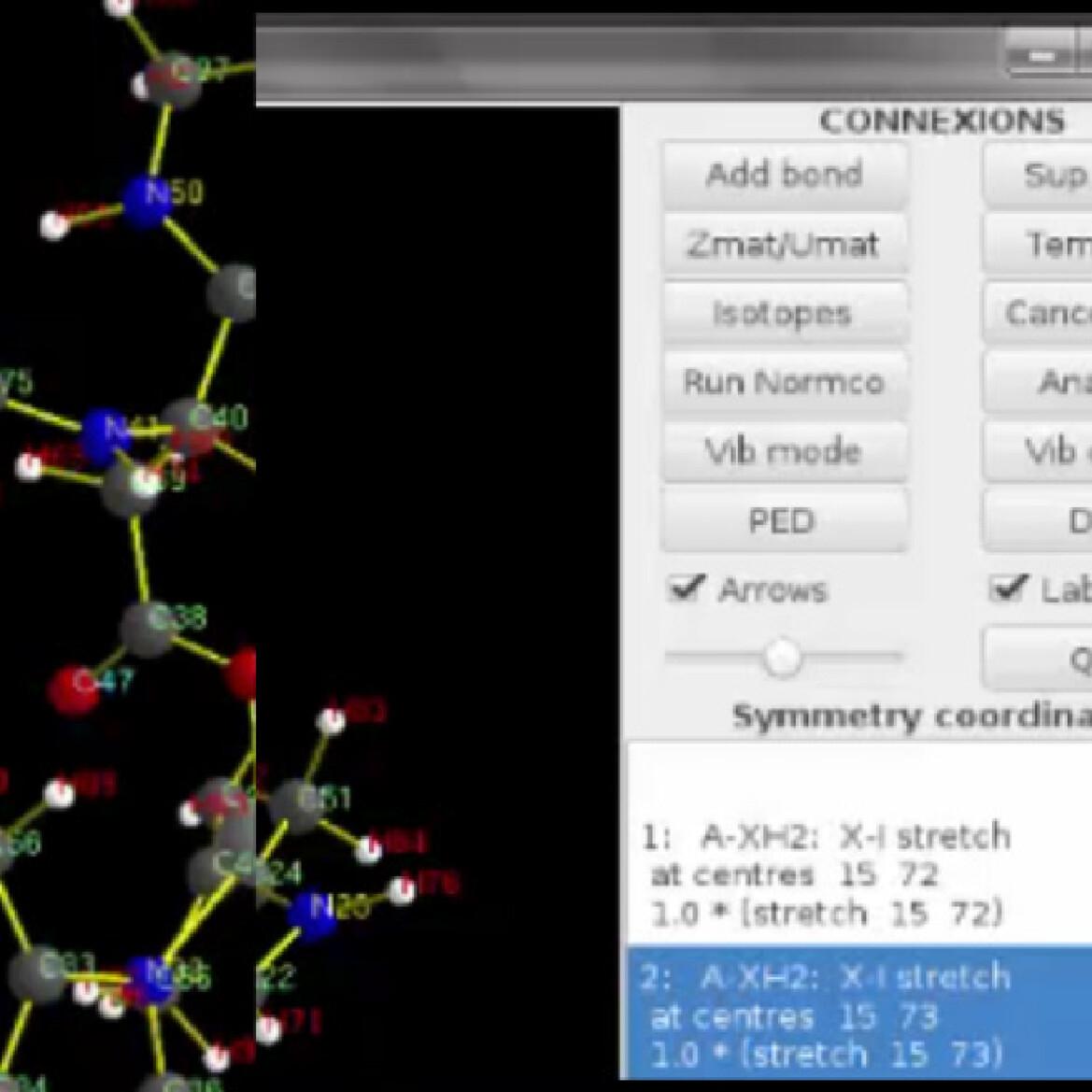
The development of infrared difference spectroscopy provides unprecedented insights on structures of complex molecules like metalloproteins. However, the relevant information can be hard to find among the many bands of the vibrational spectra. The ab initio modeling is very helpful to assign the frequencies to vibrational modes but it is a challenge to process the huge quantity of data into descriptors useful for experimentalists. To this end, we developed a new tool called VIBMOL allowing to analyze vibrational modes of molecules from hessian matrices calculated with common quantum chemistry codes. VIBMOL program runs on Unix machines. Through a new graphical interface, the users can calculate the normal modes of molecules, visualize them, simulate infrared spectra, and explore the Potential Energy Distribution of normal modes among any set of vibration coordinates. It is combined with an interface program (gosdmu) formatting relevant data from the GAUSSIAN program. VIBMOL code is available upon request to the authors. A discussion is provided to help the readers to choose between a large choice of different software and it shows how VIBMOL can make the IR assignment easier in the context of collaborations with experimentalists.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
