Michael O. Kalinkin, Dina G. Kellerman and Nadezhda I. Medvedeva
{"title":"硼磷酸盐稳定性和四极耦合常数的 Ab initio 研究","authors":"Michael O. Kalinkin, Dina G. Kellerman and Nadezhda I. Medvedeva","doi":"10.1039/D4DT01429D","DOIUrl":null,"url":null,"abstract":"<p >The DFT method was used to predict the formation energies and quadrupole coupling constants <em>C</em><small><sub>Q</sub></small> in a series of borophosphates: Li<small><sub>3</sub></small>BP<small><sub>2</sub></small>O<small><sub>8</sub></small>, Li<small><sub>2</sub></small>NaBP<small><sub>2</sub></small>O<small><sub>8</sub></small>, Na<small><sub>3</sub></small>BP<small><sub>2</sub></small>O<small><sub>8</sub></small>, Li<small><sub>2</sub></small>B<small><sub>3</sub></small>PO<small><sub>8</sub></small>, Na<small><sub>5</sub></small>B<small><sub>2</sub></small>P<small><sub>3</sub></small>O<small><sub>13</sub></small>, LiNa<small><sub>2</sub></small>B<small><sub>5</sub></small>P<small><sub>2</sub></small>O<small><sub>14</sub></small> and Na<small><sub>3</sub></small>B<small><sub>6</sub></small>PO<small><sub>13</sub></small> composed of different networks and different amounts of borate and phosphate units. The change in formation energies with increasing number of B atoms in this series is attributed to the multiplicity of boron sites and is explained by density of states calculations. The calculated <em>C</em><small><sub>Q</sub></small> values of <small><sup>7</sup></small>Li, <small><sup>23</sup></small>Na and <small><sup>11</sup></small>B are correlated with the coordination and distortion of polyhedra to elucidate the influence of local and more distant environments. As for the <em>C</em><small><sub>Q</sub></small> of <small><sup>11</sup></small>B, it should be in the ranges of 0.26–0.36, 0.48–0.84 and ∼1 MHz for boron tetrahedral distortion indices of 0.004–0.013, 0.015–0.019 and 0.033, respectively, whereas <em>C</em><small><sub>Q</sub></small> ∼3.0 MHz corresponds to boron in a triangular site. The obtained numerical relationships make it possible to predict the quadrupole frequencies for these nuclei based only on their local environment, and <em>vice versa</em>, to propose structural models from NMR data. These results provide guidance for studying similar characteristics of other borophosphates, the structure of which varies depending on the initial reaction, composition and temperature.</p>","PeriodicalId":71,"journal":{"name":"Dalton Transactions","volume":" 28","pages":" 11928-11937"},"PeriodicalIF":3.3000,"publicationDate":"2024-06-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Ab initio study of stability and quadrupole coupling constants in borophosphates\",\"authors\":\"Michael O. Kalinkin, Dina G. Kellerman and Nadezhda I. Medvedeva\",\"doi\":\"10.1039/D4DT01429D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The DFT method was used to predict the formation energies and quadrupole coupling constants <em>C</em><small><sub>Q</sub></small> in a series of borophosphates: Li<small><sub>3</sub></small>BP<small><sub>2</sub></small>O<small><sub>8</sub></small>, Li<small><sub>2</sub></small>NaBP<small><sub>2</sub></small>O<small><sub>8</sub></small>, Na<small><sub>3</sub></small>BP<small><sub>2</sub></small>O<small><sub>8</sub></small>, Li<small><sub>2</sub></small>B<small><sub>3</sub></small>PO<small><sub>8</sub></small>, Na<small><sub>5</sub></small>B<small><sub>2</sub></small>P<small><sub>3</sub></small>O<small><sub>13</sub></small>, LiNa<small><sub>2</sub></small>B<small><sub>5</sub></small>P<small><sub>2</sub></small>O<small><sub>14</sub></small> and Na<small><sub>3</sub></small>B<small><sub>6</sub></small>PO<small><sub>13</sub></small> composed of different networks and different amounts of borate and phosphate units. The change in formation energies with increasing number of B atoms in this series is attributed to the multiplicity of boron sites and is explained by density of states calculations. The calculated <em>C</em><small><sub>Q</sub></small> values of <small><sup>7</sup></small>Li, <small><sup>23</sup></small>Na and <small><sup>11</sup></small>B are correlated with the coordination and distortion of polyhedra to elucidate the influence of local and more distant environments. As for the <em>C</em><small><sub>Q</sub></small> of <small><sup>11</sup></small>B, it should be in the ranges of 0.26–0.36, 0.48–0.84 and ∼1 MHz for boron tetrahedral distortion indices of 0.004–0.013, 0.015–0.019 and 0.033, respectively, whereas <em>C</em><small><sub>Q</sub></small> ∼3.0 MHz corresponds to boron in a triangular site. The obtained numerical relationships make it possible to predict the quadrupole frequencies for these nuclei based only on their local environment, and <em>vice versa</em>, to propose structural models from NMR data. These results provide guidance for studying similar characteristics of other borophosphates, the structure of which varies depending on the initial reaction, composition and temperature.</p>\",\"PeriodicalId\":71,\"journal\":{\"name\":\"Dalton Transactions\",\"volume\":\" 28\",\"pages\":\" 11928-11937\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-06-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Dalton Transactions\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/dt/d4dt01429d\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Dalton Transactions","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dt/d4dt01429d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
Ab initio study of stability and quadrupole coupling constants in borophosphates
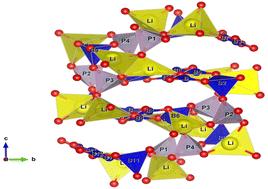
The DFT method was used to predict the formation energies and quadrupole coupling constants CQ in a series of borophosphates: Li3BP2O8, Li2NaBP2O8, Na3BP2O8, Li2B3PO8, Na5B2P3O13, LiNa2B5P2O14 and Na3B6PO13 composed of different networks and different amounts of borate and phosphate units. The change in formation energies with increasing number of B atoms in this series is attributed to the multiplicity of boron sites and is explained by density of states calculations. The calculated CQ values of 7Li, 23Na and 11B are correlated with the coordination and distortion of polyhedra to elucidate the influence of local and more distant environments. As for the CQ of 11B, it should be in the ranges of 0.26–0.36, 0.48–0.84 and ∼1 MHz for boron tetrahedral distortion indices of 0.004–0.013, 0.015–0.019 and 0.033, respectively, whereas CQ ∼3.0 MHz corresponds to boron in a triangular site. The obtained numerical relationships make it possible to predict the quadrupole frequencies for these nuclei based only on their local environment, and vice versa, to propose structural models from NMR data. These results provide guidance for studying similar characteristics of other borophosphates, the structure of which varies depending on the initial reaction, composition and temperature.
期刊介绍:
Dalton Transactions is a journal for all areas of inorganic chemistry, which encompasses the organometallic, bioinorganic and materials chemistry of the elements, with applications including synthesis, catalysis, energy conversion/storage, electrical devices and medicine. Dalton Transactions welcomes high-quality, original submissions in all of these areas and more, where the advancement of knowledge in inorganic chemistry is significant.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
