Dongsheng Zhao, Siyu Chen, Yangshuo Han, Guanqing Liu, Jinyu Liu, Qingqing Yang, Tao Zhang, Jilei Shen, Xiaolei Fan, Changquan Zhang, Tao Zhang, Qianfeng Li, Chen Chen, Qiaoquan Liu
{"title":"CRISPR/Cas9 介导的水稻优种基因突变文库","authors":"Dongsheng Zhao, Siyu Chen, Yangshuo Han, Guanqing Liu, Jinyu Liu, Qingqing Yang, Tao Zhang, Jilei Shen, Xiaolei Fan, Changquan Zhang, Tao Zhang, Qianfeng Li, Chen Chen, Qiaoquan Liu","doi":"10.1111/pbi.14422","DOIUrl":null,"url":null,"abstract":"<p>Rice seeds are an important energy source for humans. Seed traits are difficult to observe and controlled by complex networks. Therefore, mutant libraries enriched in seed traits are vital for interpreting gene functions during seed development as well as grain yield and quality formation. Using the simple and efficient genomic editing tool, several CRISPR/Cas9-based mutant libraries have been generated in rice (Chen <i>et al</i>., <span>2022</span>; Lu <i>et al</i>., <span>2017</span>; Meng <i>et al</i>., <span>2017</span>), and other crops (Bai <i>et al</i>., <span>2020</span>; Jacobs <i>et al</i>., <span>2017</span>; Liu <i>et al</i>., <span>2020</span>). Genome-wide mutants have some disadvantages (Gaillochet <i>et al</i>., <span>2021</span>), whereas appropriate-scale mutants may help focus on the special study, such as seed traits. Thus, screening specific gene sets as targets is crucial (Liu <i>et al</i>., <span>2023</span>). Besides, traditional individual editing may be beneficial for appropriate-scale population compared with the reported pooled transformation (Liu <i>et al</i>., <span>2023</span>), and has advantages when studying genes related to seed lethality.</p><p>In this study, we first identified 3288 genes with significantly differential expression using RNA sequencing (expression in seeds and twofold than leaf, hull and inflorescence, <i>p</i> < 0.05), which were defined as seed differentially expressed genes (Table S1), which may be important for seed function. Furthermore, we identified a stringent group of 1206 genes with a strong preference for seed expression, which was defined as the seed-preferred gene (expression in seeds and 10-fold than leaf, hull and inflorescence, <i>p</i> < 0.05, Table S2), and examined their functions. They are further subdivided into two categories, the 1160 specific (I, II and III) and the 46 dominant, based on the degree of tissue-restricted expression (Table S2). However, we also included 374 genes whose expression did not meet the 10-fold threshold but were hypothesized to involve in seed development from other literatures (Table S2). We also used public databases RiceXPro, RGAP and TENOR to classify the integrated 1580 genes into three subgroups, 794 endosperm-preferred genes (OsEnP), 291 embryo-preferred genes (OsEmP) and 495 others (Tables S2 and S3). These results provided more information regarding the expression of seed-preferred genes.</p><p>As an initial attempt to establish a seed-preferred gene knockout (KO) mutant library, we chose 244 genes from the above 1580 ones, including 174 OsEnPs, 56 OsEmPs and 14 others (Table S2), covering various types of expression patterns but with an emphasis on endosperm. Besides, other 66 genes of interest were used as controls. Finally, a total of 310 genes were selected for KO trial (Table S2; Figure S1a), and their expression patterns are presented through a clustering heat map as shown in Figure 1a and Table S4 and S5.</p><p>To reduce the possibility of off-target, we used strict criteria to design the guide RNAs (gRNAs) (Figure S2) and produced 375 gRNAs (Figure S1b; Table S6–S8), of which 99 were obtained from an existed library (Lu <i>et al</i>., <span>2017</span>). Notably, to ensure the acquisition of mutants and analyse the importance of gRNA design within same one gene, two gRNAs were designed for 65 genes. Then, the mutant library was constructed by transforming the design gRNAs one by one (Figure S1c,d). A total of 2688 stable T0 transgenic seedlings were generated and genotyped, which covers all 375 gRNAs (Figure S3; Table S9). Therein, 2184 of the 2598 seedlings targeted 367 gRNAs, successfully mutated. The mutation frequency was 84.06%. The remaining 90 seedlings for the eight gRNAs had no mutations (Figure 1b). Correspondingly, 302 genes got mutants and eight genes did not get mutants (Table S9). However, the mutation rate of each gRNA differed significantly (Figures S4 and S5). Therein, 96% of gRNAs yielded 3–8 mutant seedlings (Figure S6). All the mutation sites were caused by short sequence insertion and/or deletion (Figure S7), and the seedlings for most gRNAs contained frameshift mutations (Figure S3). Subsequently, we investigated the transmission of mutations from T0 to T1 generations in the planted T1 lines, and found that all 1002 decodable T0 transformants produced the expected genotypes in T1 lines following classic Mendelian law, and 92.74% of the 427 undecodable T0 transformants could be decoded in T1 lines might due to the generational reduction in sequence complexity (Figure S8). Most T0 transformants (72.16%) contained a single copy transgene region (T-DNA) through separation ratio analysis of hygromycin resistance gene, and it was easy to obtain transgene-free mutants (Table S11), which are essential for further using these novel germplasm resources. Off-target effects is a major concern for CRISPR/cas9 system. Therefore, the first putative off-target sites of each gRNA were screened using off-target tools, and probable off-target sites for the 18 gRNAs were sequenced in the T1 lines (off-score >0.6). No off-target mutations were observed in these selected gRNAs (Table S7).</p><p>To mine novel gene resources related to seed development, we then carefully measured several important seed traits for all these mutants. Specifically, at least 74 candidate genes were identified to have a significant influence on taste quality, such as apparent amylose content, protein content, and starch viscosity (Tables S12 and S13). For the grain appearance quality, 14 candidates were differed, including nine unknown OsEnP genes (Figure 1c; Table S12). In conclusion, a high proportion of genes or mutants (>50%) exhibited altered grain phenotypes, even within the limited scope of the investigation (Table S12), implying that the approach is effective for mining seed mutants.</p><p>Chalkiness is susceptible to genetic and environmental factors, resulting in difficulty to cloning the caused genes. Using this library, several chalky mutants from the unknown genes were successfully identified (Figure 1c), as one example by <i>Chalk3</i>/<i>LOC_Os03g45210</i> (Figure 1). The expression of <i>Chalk3</i> gene was preferentially high in the developing endosperm, with the highest in the middle stage during seed development (Figure 1d; Table S2). The <i>chalk3</i> mutant, <i>SG6280</i>, exhibited no visible differences in plant architecture or grain size (Figure 1e,f; Figure S9); however, the grain chalkiness significantly increased, with a pronounced core and belly white endosperm (Figure 1g–j). Another mutant from the <i>Chalk3</i> gene, <i>SG6281</i>, exhibited the same increased chalkiness (Figure S10). These results confirmed that <i>Chalk3</i> indeed plays a specific role in regulation of grain chalkiness. The chalky area of <i>chalk3</i> had notably different starch grains from those of wild type (Figure 1j). Total starch and protein contents were lower in <i>chalk3</i> grains, whereas soluble sugar levels were higher (Figure S11a–c). The chalkiness of <i>chalk3</i> mutants is susceptible to environmental influences (Figure S11d). RNA-sequencing analysis revealed that the <i>chalk3</i> mutation resulted in a number of differentially expressed genes (DEGs; Table S14). Additionally, these DEGs were significantly (<i>p</i> < 0.05) enriched for carbon and nitrogen metabolism, and plant hormone signal transduction (Figure S12). These results indicated that <i>chalk3</i> mutation alters the accumulation of stored substances in seeds, causing chalky endosperm, which might be involved in plant hormone, providing a new clue for regulating chalkiness (Zhao <i>et al</i>., <span>2022</span>).</p><p>In conclusion, we gave the expression of seed-preferred genes in rice, and established a seed-preferred mutant library on an appropriate scale based on CRISPR/Cas9 individual editing. As an example for mining novel genes using these mutants, we further elucidated the function of <i>Chalk3/LOC_Os03g45210</i> on the regulation of grain appearance quality. This manageable seed-preferred mutant library provides a resource for identifying unknown genes involved in seed development. And the batch approach may be feasible for gradually generating an individual gene mutant library that covers all genes.</p><p>The authors declare no competing interests.</p><p>Q. Li, C. C. and Q. Liu designed and supervised this study. D. Z., S. C., Y. H., G. L., J.L., Q.Y., T.Z. and J.S. performed the experiments. D. Z., S. C., X. F., C. Z., T. Z. and Q. L. analysed the data. D. Z., S. C. and Y. H. wrote this article. T. Z., Q. Li., C. C. and Q. Liu revised this article.</p>","PeriodicalId":221,"journal":{"name":"Plant Biotechnology Journal","volume":"22 11","pages":"3012-3014"},"PeriodicalIF":10.5000,"publicationDate":"2024-06-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/pbi.14422","citationCount":"0","resultStr":"{\"title\":\"A CRISPR/Cas9-mediated mutant library of seed-preferred genes in rice\",\"authors\":\"Dongsheng Zhao, Siyu Chen, Yangshuo Han, Guanqing Liu, Jinyu Liu, Qingqing Yang, Tao Zhang, Jilei Shen, Xiaolei Fan, Changquan Zhang, Tao Zhang, Qianfeng Li, Chen Chen, Qiaoquan Liu\",\"doi\":\"10.1111/pbi.14422\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Rice seeds are an important energy source for humans. Seed traits are difficult to observe and controlled by complex networks. Therefore, mutant libraries enriched in seed traits are vital for interpreting gene functions during seed development as well as grain yield and quality formation. Using the simple and efficient genomic editing tool, several CRISPR/Cas9-based mutant libraries have been generated in rice (Chen <i>et al</i>., <span>2022</span>; Lu <i>et al</i>., <span>2017</span>; Meng <i>et al</i>., <span>2017</span>), and other crops (Bai <i>et al</i>., <span>2020</span>; Jacobs <i>et al</i>., <span>2017</span>; Liu <i>et al</i>., <span>2020</span>). Genome-wide mutants have some disadvantages (Gaillochet <i>et al</i>., <span>2021</span>), whereas appropriate-scale mutants may help focus on the special study, such as seed traits. Thus, screening specific gene sets as targets is crucial (Liu <i>et al</i>., <span>2023</span>). Besides, traditional individual editing may be beneficial for appropriate-scale population compared with the reported pooled transformation (Liu <i>et al</i>., <span>2023</span>), and has advantages when studying genes related to seed lethality.</p><p>In this study, we first identified 3288 genes with significantly differential expression using RNA sequencing (expression in seeds and twofold than leaf, hull and inflorescence, <i>p</i> < 0.05), which were defined as seed differentially expressed genes (Table S1), which may be important for seed function. Furthermore, we identified a stringent group of 1206 genes with a strong preference for seed expression, which was defined as the seed-preferred gene (expression in seeds and 10-fold than leaf, hull and inflorescence, <i>p</i> < 0.05, Table S2), and examined their functions. They are further subdivided into two categories, the 1160 specific (I, II and III) and the 46 dominant, based on the degree of tissue-restricted expression (Table S2). However, we also included 374 genes whose expression did not meet the 10-fold threshold but were hypothesized to involve in seed development from other literatures (Table S2). We also used public databases RiceXPro, RGAP and TENOR to classify the integrated 1580 genes into three subgroups, 794 endosperm-preferred genes (OsEnP), 291 embryo-preferred genes (OsEmP) and 495 others (Tables S2 and S3). These results provided more information regarding the expression of seed-preferred genes.</p><p>As an initial attempt to establish a seed-preferred gene knockout (KO) mutant library, we chose 244 genes from the above 1580 ones, including 174 OsEnPs, 56 OsEmPs and 14 others (Table S2), covering various types of expression patterns but with an emphasis on endosperm. Besides, other 66 genes of interest were used as controls. Finally, a total of 310 genes were selected for KO trial (Table S2; Figure S1a), and their expression patterns are presented through a clustering heat map as shown in Figure 1a and Table S4 and S5.</p><p>To reduce the possibility of off-target, we used strict criteria to design the guide RNAs (gRNAs) (Figure S2) and produced 375 gRNAs (Figure S1b; Table S6–S8), of which 99 were obtained from an existed library (Lu <i>et al</i>., <span>2017</span>). Notably, to ensure the acquisition of mutants and analyse the importance of gRNA design within same one gene, two gRNAs were designed for 65 genes. Then, the mutant library was constructed by transforming the design gRNAs one by one (Figure S1c,d). A total of 2688 stable T0 transgenic seedlings were generated and genotyped, which covers all 375 gRNAs (Figure S3; Table S9). Therein, 2184 of the 2598 seedlings targeted 367 gRNAs, successfully mutated. The mutation frequency was 84.06%. The remaining 90 seedlings for the eight gRNAs had no mutations (Figure 1b). Correspondingly, 302 genes got mutants and eight genes did not get mutants (Table S9). However, the mutation rate of each gRNA differed significantly (Figures S4 and S5). Therein, 96% of gRNAs yielded 3–8 mutant seedlings (Figure S6). All the mutation sites were caused by short sequence insertion and/or deletion (Figure S7), and the seedlings for most gRNAs contained frameshift mutations (Figure S3). Subsequently, we investigated the transmission of mutations from T0 to T1 generations in the planted T1 lines, and found that all 1002 decodable T0 transformants produced the expected genotypes in T1 lines following classic Mendelian law, and 92.74% of the 427 undecodable T0 transformants could be decoded in T1 lines might due to the generational reduction in sequence complexity (Figure S8). Most T0 transformants (72.16%) contained a single copy transgene region (T-DNA) through separation ratio analysis of hygromycin resistance gene, and it was easy to obtain transgene-free mutants (Table S11), which are essential for further using these novel germplasm resources. Off-target effects is a major concern for CRISPR/cas9 system. Therefore, the first putative off-target sites of each gRNA were screened using off-target tools, and probable off-target sites for the 18 gRNAs were sequenced in the T1 lines (off-score >0.6). No off-target mutations were observed in these selected gRNAs (Table S7).</p><p>To mine novel gene resources related to seed development, we then carefully measured several important seed traits for all these mutants. Specifically, at least 74 candidate genes were identified to have a significant influence on taste quality, such as apparent amylose content, protein content, and starch viscosity (Tables S12 and S13). For the grain appearance quality, 14 candidates were differed, including nine unknown OsEnP genes (Figure 1c; Table S12). In conclusion, a high proportion of genes or mutants (>50%) exhibited altered grain phenotypes, even within the limited scope of the investigation (Table S12), implying that the approach is effective for mining seed mutants.</p><p>Chalkiness is susceptible to genetic and environmental factors, resulting in difficulty to cloning the caused genes. Using this library, several chalky mutants from the unknown genes were successfully identified (Figure 1c), as one example by <i>Chalk3</i>/<i>LOC_Os03g45210</i> (Figure 1). The expression of <i>Chalk3</i> gene was preferentially high in the developing endosperm, with the highest in the middle stage during seed development (Figure 1d; Table S2). The <i>chalk3</i> mutant, <i>SG6280</i>, exhibited no visible differences in plant architecture or grain size (Figure 1e,f; Figure S9); however, the grain chalkiness significantly increased, with a pronounced core and belly white endosperm (Figure 1g–j). Another mutant from the <i>Chalk3</i> gene, <i>SG6281</i>, exhibited the same increased chalkiness (Figure S10). These results confirmed that <i>Chalk3</i> indeed plays a specific role in regulation of grain chalkiness. The chalky area of <i>chalk3</i> had notably different starch grains from those of wild type (Figure 1j). Total starch and protein contents were lower in <i>chalk3</i> grains, whereas soluble sugar levels were higher (Figure S11a–c). The chalkiness of <i>chalk3</i> mutants is susceptible to environmental influences (Figure S11d). RNA-sequencing analysis revealed that the <i>chalk3</i> mutation resulted in a number of differentially expressed genes (DEGs; Table S14). Additionally, these DEGs were significantly (<i>p</i> < 0.05) enriched for carbon and nitrogen metabolism, and plant hormone signal transduction (Figure S12). These results indicated that <i>chalk3</i> mutation alters the accumulation of stored substances in seeds, causing chalky endosperm, which might be involved in plant hormone, providing a new clue for regulating chalkiness (Zhao <i>et al</i>., <span>2022</span>).</p><p>In conclusion, we gave the expression of seed-preferred genes in rice, and established a seed-preferred mutant library on an appropriate scale based on CRISPR/Cas9 individual editing. As an example for mining novel genes using these mutants, we further elucidated the function of <i>Chalk3/LOC_Os03g45210</i> on the regulation of grain appearance quality. This manageable seed-preferred mutant library provides a resource for identifying unknown genes involved in seed development. And the batch approach may be feasible for gradually generating an individual gene mutant library that covers all genes.</p><p>The authors declare no competing interests.</p><p>Q. Li, C. C. and Q. Liu designed and supervised this study. D. Z., S. C., Y. H., G. L., J.L., Q.Y., T.Z. and J.S. performed the experiments. D. Z., S. C., X. F., C. Z., T. Z. and Q. L. analysed the data. D. Z., S. C. and Y. H. wrote this article. T. Z., Q. Li., C. C. and Q. Liu revised this article.</p>\",\"PeriodicalId\":221,\"journal\":{\"name\":\"Plant Biotechnology Journal\",\"volume\":\"22 11\",\"pages\":\"3012-3014\"},\"PeriodicalIF\":10.5000,\"publicationDate\":\"2024-06-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/pbi.14422\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Plant Biotechnology Journal\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/pbi.14422\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Plant Biotechnology Journal","FirstCategoryId":"5","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/pbi.14422","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
A CRISPR/Cas9-mediated mutant library of seed-preferred genes in rice
Rice seeds are an important energy source for humans. Seed traits are difficult to observe and controlled by complex networks. Therefore, mutant libraries enriched in seed traits are vital for interpreting gene functions during seed development as well as grain yield and quality formation. Using the simple and efficient genomic editing tool, several CRISPR/Cas9-based mutant libraries have been generated in rice (Chen et al., 2022; Lu et al., 2017; Meng et al., 2017), and other crops (Bai et al., 2020; Jacobs et al., 2017; Liu et al., 2020). Genome-wide mutants have some disadvantages (Gaillochet et al., 2021), whereas appropriate-scale mutants may help focus on the special study, such as seed traits. Thus, screening specific gene sets as targets is crucial (Liu et al., 2023). Besides, traditional individual editing may be beneficial for appropriate-scale population compared with the reported pooled transformation (Liu et al., 2023), and has advantages when studying genes related to seed lethality.
In this study, we first identified 3288 genes with significantly differential expression using RNA sequencing (expression in seeds and twofold than leaf, hull and inflorescence, p < 0.05), which were defined as seed differentially expressed genes (Table S1), which may be important for seed function. Furthermore, we identified a stringent group of 1206 genes with a strong preference for seed expression, which was defined as the seed-preferred gene (expression in seeds and 10-fold than leaf, hull and inflorescence, p < 0.05, Table S2), and examined their functions. They are further subdivided into two categories, the 1160 specific (I, II and III) and the 46 dominant, based on the degree of tissue-restricted expression (Table S2). However, we also included 374 genes whose expression did not meet the 10-fold threshold but were hypothesized to involve in seed development from other literatures (Table S2). We also used public databases RiceXPro, RGAP and TENOR to classify the integrated 1580 genes into three subgroups, 794 endosperm-preferred genes (OsEnP), 291 embryo-preferred genes (OsEmP) and 495 others (Tables S2 and S3). These results provided more information regarding the expression of seed-preferred genes.
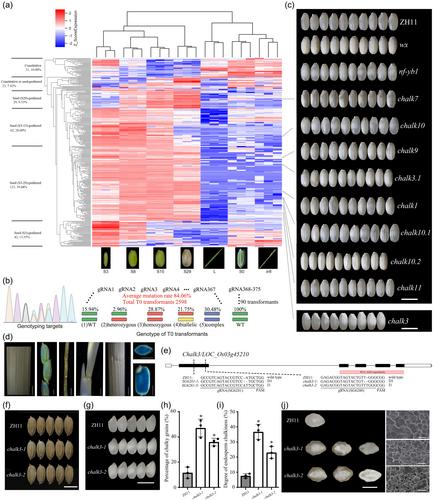
As an initial attempt to establish a seed-preferred gene knockout (KO) mutant library, we chose 244 genes from the above 1580 ones, including 174 OsEnPs, 56 OsEmPs and 14 others (Table S2), covering various types of expression patterns but with an emphasis on endosperm. Besides, other 66 genes of interest were used as controls. Finally, a total of 310 genes were selected for KO trial (Table S2; Figure S1a), and their expression patterns are presented through a clustering heat map as shown in Figure 1a and Table S4 and S5.
To reduce the possibility of off-target, we used strict criteria to design the guide RNAs (gRNAs) (Figure S2) and produced 375 gRNAs (Figure S1b; Table S6–S8), of which 99 were obtained from an existed library (Lu et al., 2017). Notably, to ensure the acquisition of mutants and analyse the importance of gRNA design within same one gene, two gRNAs were designed for 65 genes. Then, the mutant library was constructed by transforming the design gRNAs one by one (Figure S1c,d). A total of 2688 stable T0 transgenic seedlings were generated and genotyped, which covers all 375 gRNAs (Figure S3; Table S9). Therein, 2184 of the 2598 seedlings targeted 367 gRNAs, successfully mutated. The mutation frequency was 84.06%. The remaining 90 seedlings for the eight gRNAs had no mutations (Figure 1b). Correspondingly, 302 genes got mutants and eight genes did not get mutants (Table S9). However, the mutation rate of each gRNA differed significantly (Figures S4 and S5). Therein, 96% of gRNAs yielded 3–8 mutant seedlings (Figure S6). All the mutation sites were caused by short sequence insertion and/or deletion (Figure S7), and the seedlings for most gRNAs contained frameshift mutations (Figure S3). Subsequently, we investigated the transmission of mutations from T0 to T1 generations in the planted T1 lines, and found that all 1002 decodable T0 transformants produced the expected genotypes in T1 lines following classic Mendelian law, and 92.74% of the 427 undecodable T0 transformants could be decoded in T1 lines might due to the generational reduction in sequence complexity (Figure S8). Most T0 transformants (72.16%) contained a single copy transgene region (T-DNA) through separation ratio analysis of hygromycin resistance gene, and it was easy to obtain transgene-free mutants (Table S11), which are essential for further using these novel germplasm resources. Off-target effects is a major concern for CRISPR/cas9 system. Therefore, the first putative off-target sites of each gRNA were screened using off-target tools, and probable off-target sites for the 18 gRNAs were sequenced in the T1 lines (off-score >0.6). No off-target mutations were observed in these selected gRNAs (Table S7).
To mine novel gene resources related to seed development, we then carefully measured several important seed traits for all these mutants. Specifically, at least 74 candidate genes were identified to have a significant influence on taste quality, such as apparent amylose content, protein content, and starch viscosity (Tables S12 and S13). For the grain appearance quality, 14 candidates were differed, including nine unknown OsEnP genes (Figure 1c; Table S12). In conclusion, a high proportion of genes or mutants (>50%) exhibited altered grain phenotypes, even within the limited scope of the investigation (Table S12), implying that the approach is effective for mining seed mutants.
Chalkiness is susceptible to genetic and environmental factors, resulting in difficulty to cloning the caused genes. Using this library, several chalky mutants from the unknown genes were successfully identified (Figure 1c), as one example by Chalk3/LOC_Os03g45210 (Figure 1). The expression of Chalk3 gene was preferentially high in the developing endosperm, with the highest in the middle stage during seed development (Figure 1d; Table S2). The chalk3 mutant, SG6280, exhibited no visible differences in plant architecture or grain size (Figure 1e,f; Figure S9); however, the grain chalkiness significantly increased, with a pronounced core and belly white endosperm (Figure 1g–j). Another mutant from the Chalk3 gene, SG6281, exhibited the same increased chalkiness (Figure S10). These results confirmed that Chalk3 indeed plays a specific role in regulation of grain chalkiness. The chalky area of chalk3 had notably different starch grains from those of wild type (Figure 1j). Total starch and protein contents were lower in chalk3 grains, whereas soluble sugar levels were higher (Figure S11a–c). The chalkiness of chalk3 mutants is susceptible to environmental influences (Figure S11d). RNA-sequencing analysis revealed that the chalk3 mutation resulted in a number of differentially expressed genes (DEGs; Table S14). Additionally, these DEGs were significantly (p < 0.05) enriched for carbon and nitrogen metabolism, and plant hormone signal transduction (Figure S12). These results indicated that chalk3 mutation alters the accumulation of stored substances in seeds, causing chalky endosperm, which might be involved in plant hormone, providing a new clue for regulating chalkiness (Zhao et al., 2022).
In conclusion, we gave the expression of seed-preferred genes in rice, and established a seed-preferred mutant library on an appropriate scale based on CRISPR/Cas9 individual editing. As an example for mining novel genes using these mutants, we further elucidated the function of Chalk3/LOC_Os03g45210 on the regulation of grain appearance quality. This manageable seed-preferred mutant library provides a resource for identifying unknown genes involved in seed development. And the batch approach may be feasible for gradually generating an individual gene mutant library that covers all genes.
The authors declare no competing interests.
Q. Li, C. C. and Q. Liu designed and supervised this study. D. Z., S. C., Y. H., G. L., J.L., Q.Y., T.Z. and J.S. performed the experiments. D. Z., S. C., X. F., C. Z., T. Z. and Q. L. analysed the data. D. Z., S. C. and Y. H. wrote this article. T. Z., Q. Li., C. C. and Q. Liu revised this article.
期刊介绍:
Plant Biotechnology Journal aspires to publish original research and insightful reviews of high impact, authored by prominent researchers in applied plant science. The journal places a special emphasis on molecular plant sciences and their practical applications through plant biotechnology. Our goal is to establish a platform for showcasing significant advances in the field, encompassing curiosity-driven studies with potential applications, strategic research in plant biotechnology, scientific analysis of crucial issues for the beneficial utilization of plant sciences, and assessments of the performance of plant biotechnology products in practical applications.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
