Abdallah Abou Hajal, Richard A. Bryce, Boulbaba Ben Amor, Noor Atatreh and Mohammad A. Ghattas*,
{"title":"利用 BAD 分子过滤器提高胶体聚集小分子检测的准确性和化学空间覆盖率。","authors":"Abdallah Abou Hajal, Richard A. Bryce, Boulbaba Ben Amor, Noor Atatreh and Mohammad A. Ghattas*, ","doi":"10.1021/acs.jcim.4c00363","DOIUrl":null,"url":null,"abstract":"<p >The ability to conduct effective high throughput screening (HTS) campaigns in drug discovery is often hampered by the detection of false positives in these assays due to small colloidally aggregating molecules (SCAMs). SCAMs can produce artifactual hits in HTS by nonspecific inhibition of the protein target. In this work, we present a new computational prediction tool for detecting SCAMs based on their 2D chemical structure. The tool, called the boosted aggregation detection (BAD) molecule filter, employs decision tree ensemble methods, namely, the CatBoost classifier and the light gradient-boosting machine, to significantly improve the detection of SCAMs. In developing the filter, we explore models trained on individual data sets, a consensus approach using these models, and, third, a merged data set approach, each tailored for specific drug discovery needs. The individual data set method emerged as most effective, achieving 93% sensitivity and 90% specificity, outperforming existing state-of-the-art models by 20 and 5%, respectively. The consensus models offer broader chemical space coverage, exceeding 90% for all testing sets. This feature is an important aspect particularly for early stage medicinal chemistry projects, and provides information on applicability domain. Meanwhile, the merged data set models demonstrated robust performance, with a notable sensitivity of 79% in the comprehensive 10-fold cross-validation test set. A SHAP analysis of model features indicates the importance of hydrophobicity and molecular complexity as primary factors influencing the aggregation propensity. The BAD molecule filter is readily accessible for the public usage on https://molmodlab-aau.com/Tools.html. This filter provides a new, more robust tool for aggregate prediction in the early stages of drug discovery to optimize hit rates and reduce associated testing and validation overheads.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 13","pages":"4991–5005"},"PeriodicalIF":5.3000,"publicationDate":"2024-06-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Boosting the Accuracy and Chemical Space Coverage of the Detection of Small Colloidal Aggregating Molecules Using the BAD Molecule Filter\",\"authors\":\"Abdallah Abou Hajal, Richard A. Bryce, Boulbaba Ben Amor, Noor Atatreh and Mohammad A. Ghattas*, \",\"doi\":\"10.1021/acs.jcim.4c00363\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The ability to conduct effective high throughput screening (HTS) campaigns in drug discovery is often hampered by the detection of false positives in these assays due to small colloidally aggregating molecules (SCAMs). SCAMs can produce artifactual hits in HTS by nonspecific inhibition of the protein target. In this work, we present a new computational prediction tool for detecting SCAMs based on their 2D chemical structure. The tool, called the boosted aggregation detection (BAD) molecule filter, employs decision tree ensemble methods, namely, the CatBoost classifier and the light gradient-boosting machine, to significantly improve the detection of SCAMs. In developing the filter, we explore models trained on individual data sets, a consensus approach using these models, and, third, a merged data set approach, each tailored for specific drug discovery needs. The individual data set method emerged as most effective, achieving 93% sensitivity and 90% specificity, outperforming existing state-of-the-art models by 20 and 5%, respectively. The consensus models offer broader chemical space coverage, exceeding 90% for all testing sets. This feature is an important aspect particularly for early stage medicinal chemistry projects, and provides information on applicability domain. Meanwhile, the merged data set models demonstrated robust performance, with a notable sensitivity of 79% in the comprehensive 10-fold cross-validation test set. A SHAP analysis of model features indicates the importance of hydrophobicity and molecular complexity as primary factors influencing the aggregation propensity. The BAD molecule filter is readily accessible for the public usage on https://molmodlab-aau.com/Tools.html. This filter provides a new, more robust tool for aggregate prediction in the early stages of drug discovery to optimize hit rates and reduce associated testing and validation overheads.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"64 13\",\"pages\":\"4991–5005\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2024-06-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00363\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00363","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
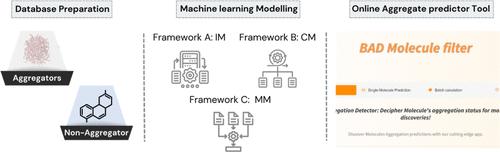
Boosting the Accuracy and Chemical Space Coverage of the Detection of Small Colloidal Aggregating Molecules Using the BAD Molecule Filter
The ability to conduct effective high throughput screening (HTS) campaigns in drug discovery is often hampered by the detection of false positives in these assays due to small colloidally aggregating molecules (SCAMs). SCAMs can produce artifactual hits in HTS by nonspecific inhibition of the protein target. In this work, we present a new computational prediction tool for detecting SCAMs based on their 2D chemical structure. The tool, called the boosted aggregation detection (BAD) molecule filter, employs decision tree ensemble methods, namely, the CatBoost classifier and the light gradient-boosting machine, to significantly improve the detection of SCAMs. In developing the filter, we explore models trained on individual data sets, a consensus approach using these models, and, third, a merged data set approach, each tailored for specific drug discovery needs. The individual data set method emerged as most effective, achieving 93% sensitivity and 90% specificity, outperforming existing state-of-the-art models by 20 and 5%, respectively. The consensus models offer broader chemical space coverage, exceeding 90% for all testing sets. This feature is an important aspect particularly for early stage medicinal chemistry projects, and provides information on applicability domain. Meanwhile, the merged data set models demonstrated robust performance, with a notable sensitivity of 79% in the comprehensive 10-fold cross-validation test set. A SHAP analysis of model features indicates the importance of hydrophobicity and molecular complexity as primary factors influencing the aggregation propensity. The BAD molecule filter is readily accessible for the public usage on https://molmodlab-aau.com/Tools.html. This filter provides a new, more robust tool for aggregate prediction in the early stages of drug discovery to optimize hit rates and reduce associated testing and validation overheads.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.




 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
