Anna Kennedy, Ella Richardson, Jonathan Higham, Panagiotis Kotsantis, Richard Mort, Barbara Bo-Ju Shih
{"title":"Evergene:以基因为中心对原发性肿瘤进行大规模分析的交互式网络工具。","authors":"Anna Kennedy, Ella Richardson, Jonathan Higham, Panagiotis Kotsantis, Richard Mort, Barbara Bo-Ju Shih","doi":"10.1093/bioadv/vbae092","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>The data sharing of large comprehensive cancer research projects, such as The Cancer Genome Atlas (TCGA), has improved the availability of high-quality data to research labs around the world. However, due to the volume and inherent complexity of high-throughput omics data, analysis of this is limited by the capacity for performing data processing through programming languages such as R or Python. Existing webtools lack functionality that supports large-scale analysis; typically, users can only input one gene, or a gene list condensed into a gene set, instead of individual gene-level analysis. Furthermore, analysis results are usually displayed without other sample-level molecular or clinical annotations. To address these gaps in the existing webtools, we have developed Evergene using R and Shiny.</p><p><strong>Results: </strong>Evergene is a user-friendly webtool that utilizes RNA-sequencing data, alongside other sample and clinical annotation, for large-scale gene-centric analysis, including principal component analysis (PCA), survival analysis (SA), and correlation analysis (CA). Moreover, Evergene achieves in-depth analysis of cancer transcriptomic data which can be explored through dimensional reduction methods, relating gene expression with clinical events or other sample information, such as ethnicity, histological classification, and molecular indices. Lastly, users can upload custom data to Evergene for analysis.</p><p><strong>Availability and implementation: </strong>Evergene webtool is available at https://bshihlab.shinyapps.io/evergene/. The source code and example user input dataset are available at https://github.com/bshihlab/evergene.</p>","PeriodicalId":72368,"journal":{"name":"Bioinformatics advances","volume":"4 1","pages":"vbae092"},"PeriodicalIF":2.8000,"publicationDate":"2024-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11213629/pdf/","citationCount":"0","resultStr":"{\"title\":\"Evergene: an interactive webtool for large-scale gene-centric analysis of primary tumours.\",\"authors\":\"Anna Kennedy, Ella Richardson, Jonathan Higham, Panagiotis Kotsantis, Richard Mort, Barbara Bo-Ju Shih\",\"doi\":\"10.1093/bioadv/vbae092\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>The data sharing of large comprehensive cancer research projects, such as The Cancer Genome Atlas (TCGA), has improved the availability of high-quality data to research labs around the world. However, due to the volume and inherent complexity of high-throughput omics data, analysis of this is limited by the capacity for performing data processing through programming languages such as R or Python. Existing webtools lack functionality that supports large-scale analysis; typically, users can only input one gene, or a gene list condensed into a gene set, instead of individual gene-level analysis. Furthermore, analysis results are usually displayed without other sample-level molecular or clinical annotations. To address these gaps in the existing webtools, we have developed Evergene using R and Shiny.</p><p><strong>Results: </strong>Evergene is a user-friendly webtool that utilizes RNA-sequencing data, alongside other sample and clinical annotation, for large-scale gene-centric analysis, including principal component analysis (PCA), survival analysis (SA), and correlation analysis (CA). Moreover, Evergene achieves in-depth analysis of cancer transcriptomic data which can be explored through dimensional reduction methods, relating gene expression with clinical events or other sample information, such as ethnicity, histological classification, and molecular indices. Lastly, users can upload custom data to Evergene for analysis.</p><p><strong>Availability and implementation: </strong>Evergene webtool is available at https://bshihlab.shinyapps.io/evergene/. The source code and example user input dataset are available at https://github.com/bshihlab/evergene.</p>\",\"PeriodicalId\":72368,\"journal\":{\"name\":\"Bioinformatics advances\",\"volume\":\"4 1\",\"pages\":\"vbae092\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-06-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11213629/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics advances\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/bioadv/vbae092\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/bioadv/vbae092","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
动机癌症基因组图谱(TCGA)等大型综合癌症研究项目的数据共享,提高了世界各地研究实验室对高质量数据的可用性。然而,由于高通量 omics 数据的数量和固有的复杂性,通过 R 或 Python 等编程语言进行数据处理的能力限制了对这些数据的分析。现有的网络工具缺乏支持大规模分析的功能;通常情况下,用户只能输入一个基因或浓缩成一个基因集的基因列表,而不能进行单个基因层面的分析。此外,分析结果的显示通常没有其他样本级分子或临床注释。为了填补现有网络工具的这些空白,我们使用 R 和 Shiny.Results 开发了 Evergene:Evergene是一个用户友好型网络工具,它利用RNA测序数据以及其他样本和临床注释,进行以基因为中心的大规模分析,包括主成分分析(PCA)、生存分析(SA)和相关分析(CA)。此外,Evergene 还能对癌症转录组数据进行深入分析,并通过降维方法将基因表达与临床事件或其他样本信息(如种族、组织学分类和分子指数)联系起来。最后,用户还可以将自定义数据上传到 Evergene 进行分析:Evergene 网络工具可从 https://bshihlab.shinyapps.io/evergene/ 网站获取。源代码和用户输入数据集示例见 https://github.com/bshihlab/evergene。
Evergene: an interactive webtool for large-scale gene-centric analysis of primary tumours.
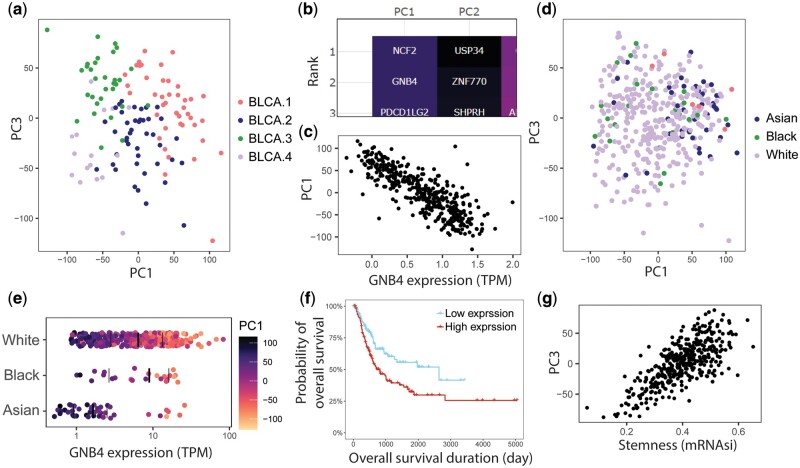
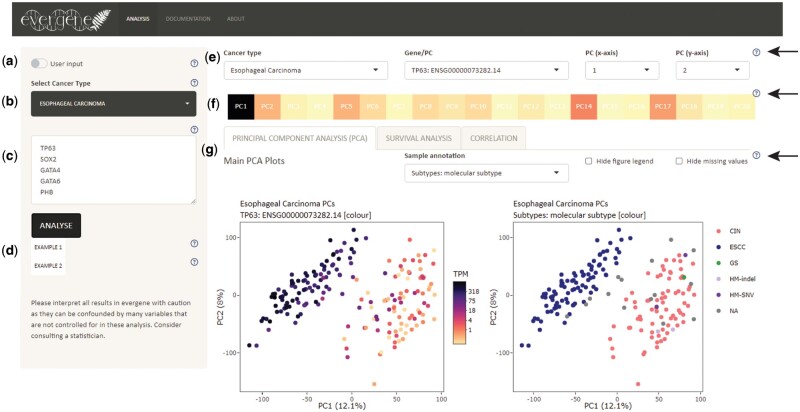
Motivation: The data sharing of large comprehensive cancer research projects, such as The Cancer Genome Atlas (TCGA), has improved the availability of high-quality data to research labs around the world. However, due to the volume and inherent complexity of high-throughput omics data, analysis of this is limited by the capacity for performing data processing through programming languages such as R or Python. Existing webtools lack functionality that supports large-scale analysis; typically, users can only input one gene, or a gene list condensed into a gene set, instead of individual gene-level analysis. Furthermore, analysis results are usually displayed without other sample-level molecular or clinical annotations. To address these gaps in the existing webtools, we have developed Evergene using R and Shiny.
Results: Evergene is a user-friendly webtool that utilizes RNA-sequencing data, alongside other sample and clinical annotation, for large-scale gene-centric analysis, including principal component analysis (PCA), survival analysis (SA), and correlation analysis (CA). Moreover, Evergene achieves in-depth analysis of cancer transcriptomic data which can be explored through dimensional reduction methods, relating gene expression with clinical events or other sample information, such as ethnicity, histological classification, and molecular indices. Lastly, users can upload custom data to Evergene for analysis.
Availability and implementation: Evergene webtool is available at https://bshihlab.shinyapps.io/evergene/. The source code and example user input dataset are available at https://github.com/bshihlab/evergene.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
