{"title":"通过基于配体的方法和前瞻性亲和力测试进行支架跳转化合物鉴定。","authors":"Itsuki Maeda, Shunsuke Tamura, Yoshihiro Ogura, Takayuki Serizawa, Takashi Shimada, Ryo Kunimoto, Tomoyuki Miyao","doi":"10.1021/acs.jcim.4c00342","DOIUrl":null,"url":null,"abstract":"<p><p>Scaffold-hopped (SH) compounds are bioactive compounds structurally different from known active compounds. Identifying SH compounds in the ligand-based approaches has been a central issue in medicinal chemistry, and various molecular representations of scaffold hopping have been proposed. However, appropriate representations for SH compound identification remain unclear. Herein, the ability of SH compound identification among several representations was fairly evaluated based on retrospective validation and prospective demonstration. In the retrospective validation, the combinations of two screening algorithms and four two- and three-dimensional molecular representations were compared using controlled data sets for the early identification of SH compounds. We found that the combination of the support vector machine and extended connectivity fingerprint with bond diameter 4 (SVM-ECFP4) and SVM and the rapid overlay of chemical structures (SVM-ROCS) showed a relatively high performance. The compounds that were highly ranked by SVM-ROCS did not share substructures with the active training compounds, while those ranked by SVM-ECFP4 were mostly recombinant. In the prospective demonstration, 93 SH compounds were prepared by screening the Namiki database using SVM-ROCS, targeting ABL1 inhibitors. The primary screening using surface plasmon resonance suggested five active compounds; however, in the competitive binding assays with adenosine triphosphate, no hits were found.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"5557-5569"},"PeriodicalIF":5.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11267578/pdf/","citationCount":"0","resultStr":"{\"title\":\"Scaffold-Hopped Compound Identification by Ligand-Based Approaches with a Prospective Affinity Test.\",\"authors\":\"Itsuki Maeda, Shunsuke Tamura, Yoshihiro Ogura, Takayuki Serizawa, Takashi Shimada, Ryo Kunimoto, Tomoyuki Miyao\",\"doi\":\"10.1021/acs.jcim.4c00342\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Scaffold-hopped (SH) compounds are bioactive compounds structurally different from known active compounds. Identifying SH compounds in the ligand-based approaches has been a central issue in medicinal chemistry, and various molecular representations of scaffold hopping have been proposed. However, appropriate representations for SH compound identification remain unclear. Herein, the ability of SH compound identification among several representations was fairly evaluated based on retrospective validation and prospective demonstration. In the retrospective validation, the combinations of two screening algorithms and four two- and three-dimensional molecular representations were compared using controlled data sets for the early identification of SH compounds. We found that the combination of the support vector machine and extended connectivity fingerprint with bond diameter 4 (SVM-ECFP4) and SVM and the rapid overlay of chemical structures (SVM-ROCS) showed a relatively high performance. The compounds that were highly ranked by SVM-ROCS did not share substructures with the active training compounds, while those ranked by SVM-ECFP4 were mostly recombinant. In the prospective demonstration, 93 SH compounds were prepared by screening the Namiki database using SVM-ROCS, targeting ABL1 inhibitors. The primary screening using surface plasmon resonance suggested five active compounds; however, in the competitive binding assays with adenosine triphosphate, no hits were found.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\" \",\"pages\":\"5557-5569\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2024-07-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11267578/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jcim.4c00342\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/7/1 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00342","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/1 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
摘要
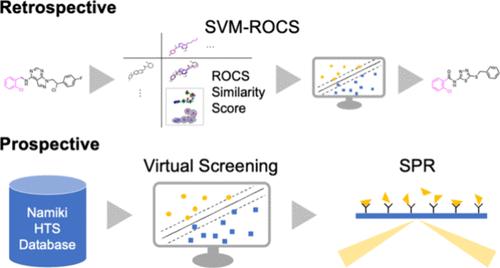
支架跳跃(SH)化合物是在结构上不同于已知活性化合物的生物活性化合物。在基于配体的方法中识别 SH 化合物一直是药物化学领域的核心问题,人们提出了各种支架跳跃的分子表示方法。然而,用于 SH 化合物鉴定的适当表征仍不明确。在此,我们通过回顾性验证和前瞻性论证,对几种表征之间的SH化合物鉴定能力进行了公正的评估。在回顾性验证中,使用对照数据集比较了两种筛选算法和四种二维和三维分子表征的组合,以早期识别 SH 化合物。我们发现,支持向量机和具有键直径 4 的扩展连通性指纹(SVM-ECFP4)以及 SVM 和化学结构快速叠加(SVM-ROCS)的组合显示出相对较高的性能。SVM-ROCS 排名靠前的化合物与活性训练化合物不共享子结构,而 SVM-ECFP4 排名靠前的化合物大多是重组化合物。在前瞻性演示中,通过使用 SVM-ROCS 对并木数据库进行筛选,制备了 93 个 SH 化合物,目标是 ABL1 抑制剂。利用表面等离子共振进行的初筛提出了五个活性化合物;但是,在与三磷酸腺苷的竞争性结合试验中,没有发现任何命中化合物。
Scaffold-Hopped Compound Identification by Ligand-Based Approaches with a Prospective Affinity Test.
Scaffold-hopped (SH) compounds are bioactive compounds structurally different from known active compounds. Identifying SH compounds in the ligand-based approaches has been a central issue in medicinal chemistry, and various molecular representations of scaffold hopping have been proposed. However, appropriate representations for SH compound identification remain unclear. Herein, the ability of SH compound identification among several representations was fairly evaluated based on retrospective validation and prospective demonstration. In the retrospective validation, the combinations of two screening algorithms and four two- and three-dimensional molecular representations were compared using controlled data sets for the early identification of SH compounds. We found that the combination of the support vector machine and extended connectivity fingerprint with bond diameter 4 (SVM-ECFP4) and SVM and the rapid overlay of chemical structures (SVM-ROCS) showed a relatively high performance. The compounds that were highly ranked by SVM-ROCS did not share substructures with the active training compounds, while those ranked by SVM-ECFP4 were mostly recombinant. In the prospective demonstration, 93 SH compounds were prepared by screening the Namiki database using SVM-ROCS, targeting ABL1 inhibitors. The primary screening using surface plasmon resonance suggested five active compounds; however, in the competitive binding assays with adenosine triphosphate, no hits were found.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
