{"title":"利用 NanoStrand-seq 在染色体尺度上同时进行基因变异的从头调用和分期。","authors":"Xiuzhen Bai, Zonggui Chen, Kexuan Chen, Zixin Wu, Rui Wang, Jun'e Liu, Liang Chang, Lu Wen, Fuchou Tang","doi":"10.1038/s41421-024-00694-9","DOIUrl":null,"url":null,"abstract":"<p><p>The successful accomplishment of the first telomere-to-telomere human genome assembly, T2T-CHM13, marked a milestone in achieving completeness of the human reference genome. The upcoming era of genome study will focus on fully phased diploid genome assembly, with an emphasis on genetic differences between individual haplotypes. Most existing sequencing approaches only achieved localized haplotype phasing and relied on additional pedigree information for further whole-chromosome scale phasing. The short-read-based Strand-seq method is able to directly phase single nucleotide polymorphisms (SNPs) at whole-chromosome scale but falls short when it comes to phasing structural variations (SVs). To shed light on this issue, we developed a Nanopore sequencing platform-based Strand-seq approach, which we named NanoStrand-seq. This method allowed for de novo SNP calling with high precision (99.52%) and acheived a superior phasing accuracy (0.02% Hamming error rate) at whole-chromosome scale, a level of performance comparable to Strand-seq for haplotype phasing of the GM12878 genome. Importantly, we demonstrated that NanoStrand-seq can efficiently resolve the MHC locus, a highly polymorphic genomic region. Moreover, NanoStrand-seq enabled independent direct calling and phasing of deletions and insertions at whole-chromosome level; when applied to long genomic regions of SNP homozygosity, it outperformed the strategy that combined Strand-seq with bulk long-read sequencing. Finally, we showed that, like Strand-seq, NanoStrand-seq was also applicable to primary cultured cells. Together, here we provided a novel methodology that enabled interrogation of a full spectrum of haplotype-resolved SNPs and SVs at whole-chromosome scale, with broad applications for species with diploid or even potentially polypoid genomes.</p>","PeriodicalId":9674,"journal":{"name":"Cell Discovery","volume":"10 1","pages":"74"},"PeriodicalIF":12.5000,"publicationDate":"2024-07-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11231365/pdf/","citationCount":"0","resultStr":"{\"title\":\"Simultaneous de novo calling and phasing of genetic variants at chromosome-scale using NanoStrand-seq.\",\"authors\":\"Xiuzhen Bai, Zonggui Chen, Kexuan Chen, Zixin Wu, Rui Wang, Jun'e Liu, Liang Chang, Lu Wen, Fuchou Tang\",\"doi\":\"10.1038/s41421-024-00694-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The successful accomplishment of the first telomere-to-telomere human genome assembly, T2T-CHM13, marked a milestone in achieving completeness of the human reference genome. The upcoming era of genome study will focus on fully phased diploid genome assembly, with an emphasis on genetic differences between individual haplotypes. Most existing sequencing approaches only achieved localized haplotype phasing and relied on additional pedigree information for further whole-chromosome scale phasing. The short-read-based Strand-seq method is able to directly phase single nucleotide polymorphisms (SNPs) at whole-chromosome scale but falls short when it comes to phasing structural variations (SVs). To shed light on this issue, we developed a Nanopore sequencing platform-based Strand-seq approach, which we named NanoStrand-seq. This method allowed for de novo SNP calling with high precision (99.52%) and acheived a superior phasing accuracy (0.02% Hamming error rate) at whole-chromosome scale, a level of performance comparable to Strand-seq for haplotype phasing of the GM12878 genome. Importantly, we demonstrated that NanoStrand-seq can efficiently resolve the MHC locus, a highly polymorphic genomic region. Moreover, NanoStrand-seq enabled independent direct calling and phasing of deletions and insertions at whole-chromosome level; when applied to long genomic regions of SNP homozygosity, it outperformed the strategy that combined Strand-seq with bulk long-read sequencing. Finally, we showed that, like Strand-seq, NanoStrand-seq was also applicable to primary cultured cells. Together, here we provided a novel methodology that enabled interrogation of a full spectrum of haplotype-resolved SNPs and SVs at whole-chromosome scale, with broad applications for species with diploid or even potentially polypoid genomes.</p>\",\"PeriodicalId\":9674,\"journal\":{\"name\":\"Cell Discovery\",\"volume\":\"10 1\",\"pages\":\"74\"},\"PeriodicalIF\":12.5000,\"publicationDate\":\"2024-07-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11231365/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cell Discovery\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1038/s41421-024-00694-9\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Discovery","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41421-024-00694-9","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
第一个端粒到端粒人类基因组T2T-CHM13的成功组装标志着人类参考基因组完整性的一个里程碑。即将到来的基因组研究时代将以全期二倍体基因组组装为重点,强调个体单倍型之间的遗传差异。现有的大多数测序方法只能实现局部的单倍型分期,进一步的全染色体规模分期需要依赖额外的血统信息。基于短读数的 Strand-seq 方法能够直接对全染色体范围内的单核苷酸多态性(SNPs)进行分型,但在对结构变异(SVs)进行分型时却存在不足。为了解决这个问题,我们开发了一种基于纳米孔测序平台的Strand-seq方法,并将其命名为NanoStrand-seq。这种方法允许高精度(99.52%)的从头SNP调用,并在全染色体范围内实现了卓越的分期准确性(0.02%的汉明错误率),其性能水平与Strand-seq对GM12878基因组的单体型分期相当。重要的是,我们证明了 NanoStrand-seq 能够高效解析 MHC 基因座这一高度多态的基因组区域。此外,NanoStrand-seq 还能在全染色体水平上对缺失和插入进行独立的直接调用和分期;当应用于 SNP 同源性的长基因组区域时,它的表现优于将 Strand-seq 与批量长线程测序相结合的策略。最后,我们表明,与 Strand-seq 一样,NanoStrand-seq 也适用于原代培养细胞。总之,我们在这里提供了一种新的方法,能够在全染色体范围内对单倍型解析的 SNP 和 SV 进行全方位的检测,可广泛应用于具有二倍体甚至潜在多倍体基因组的物种。
Simultaneous de novo calling and phasing of genetic variants at chromosome-scale using NanoStrand-seq.
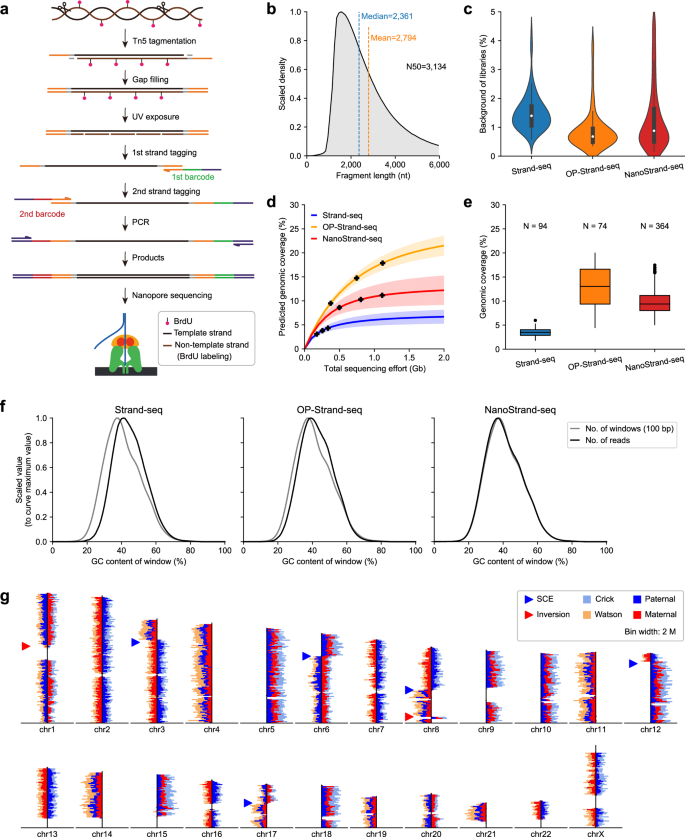
The successful accomplishment of the first telomere-to-telomere human genome assembly, T2T-CHM13, marked a milestone in achieving completeness of the human reference genome. The upcoming era of genome study will focus on fully phased diploid genome assembly, with an emphasis on genetic differences between individual haplotypes. Most existing sequencing approaches only achieved localized haplotype phasing and relied on additional pedigree information for further whole-chromosome scale phasing. The short-read-based Strand-seq method is able to directly phase single nucleotide polymorphisms (SNPs) at whole-chromosome scale but falls short when it comes to phasing structural variations (SVs). To shed light on this issue, we developed a Nanopore sequencing platform-based Strand-seq approach, which we named NanoStrand-seq. This method allowed for de novo SNP calling with high precision (99.52%) and acheived a superior phasing accuracy (0.02% Hamming error rate) at whole-chromosome scale, a level of performance comparable to Strand-seq for haplotype phasing of the GM12878 genome. Importantly, we demonstrated that NanoStrand-seq can efficiently resolve the MHC locus, a highly polymorphic genomic region. Moreover, NanoStrand-seq enabled independent direct calling and phasing of deletions and insertions at whole-chromosome level; when applied to long genomic regions of SNP homozygosity, it outperformed the strategy that combined Strand-seq with bulk long-read sequencing. Finally, we showed that, like Strand-seq, NanoStrand-seq was also applicable to primary cultured cells. Together, here we provided a novel methodology that enabled interrogation of a full spectrum of haplotype-resolved SNPs and SVs at whole-chromosome scale, with broad applications for species with diploid or even potentially polypoid genomes.
Cell DiscoveryBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
24.20
自引率
0.60%
发文量
120
审稿时长
20 weeks
期刊介绍:
Cell Discovery is a cutting-edge, open access journal published by Springer Nature in collaboration with the Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences (CAS). Our aim is to provide a dynamic and accessible platform for scientists to showcase their exceptional original research.
Cell Discovery covers a wide range of topics within the fields of molecular and cell biology. We eagerly publish results of great significance and that are of broad interest to the scientific community. With an international authorship and a focus on basic life sciences, our journal is a valued member of Springer Nature's prestigious Molecular Cell Biology journals.
In summary, Cell Discovery offers a fresh approach to scholarly publishing, enabling scientists from around the world to share their exceptional findings in molecular and cell biology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
