Christoph Dockter, Søren Knudsen, Magnus Wohlfahrt Rasmussen, Birgitte Skadhauge, Birger Lindberg Møller
{"title":"只需 FIND-IT:利用诱导突变的真正力量。","authors":"Christoph Dockter, Søren Knudsen, Magnus Wohlfahrt Rasmussen, Birgitte Skadhauge, Birger Lindberg Møller","doi":"10.1111/pbi.14427","DOIUrl":null,"url":null,"abstract":"<p>In nature, genetic variation occurs in every population and results in the evolution of a diversity of new properties, some of which promote the survival of the species. To accelerate nature's evolution based on genetic diversity, plant breeders may induce additional mutations to raise the number of genetic variations increasing the chances to obtain varieties with new desired traits like improved nutritive quality, yields and resilience to biotic and abiotic stress factors. Induced mutagenesis based on chemical mutagens is considered non-GM and has been used in barley (<i>Hordeum vulgare</i>) for decades (Hansson <i>et al</i>., <span>2024</span>). Reverse genetic techniques including TILLING (Targeting Induced Local Lesions in Genomes) screening methodology and more recently TILLING-by-sequencing spinoffs are tools used to identify individual plant variants with the desired valuable genomic alterations. However, these tools are hampered by low mutation capacity.</p><p>TILLING is a PCR-based technique designed to detect mismatched single nucleotides in a target gene. In 2023, Szarejko and her research group in Poland published a thorough overview of the TILLING success stories within the last 20 years (Szurman-Zubrzycka <i>et al</i>., <span>2023</span>) including a description of the TILLING populations in different barley cultivars and landraces obtained following chemical mutagenesis (Figure 1a). The TILLING population sizes range between 1372 and 9600 individual plant variants. The mutation frequencies are individually chosen and dose-dependent (1/154–1/2500 Kbp). When multiplied (# of individuals × mutations per individual), the total number of mutations present in barley TILLING populations ranges between 10 and 100 million (Figure 1a). This may sound like a lot, but with a barley genome size of around 4300 Mbp (here, RGT Planet; Jayakodi <i>et al</i>., <span>2020</span>), less than 2% of the nucleotides in the entire population are mutated. This severely reduces the possibility to find a desired mutation in TILLING populations. The FIND-IT technology is a new approach overriding these constraints.</p><p>The FIND-IT technology was published in Science Advances in 2022 (Figure 1b) (Knudsen <i>et al</i>., <span>2022</span>) and provides an agile and high-throughput approach to screen unprecedented large size chemically induced variant populations. FIND-IT combines systematic sample pooling and splitting with high-sensitivity, droplet digital PCR (ddPCR)–based genotyping for targeted identification of desired traits at single-nucleotide resolution. The ddPCR technology is 1000-fold more sensitive than conventional PCR. The FIND-IT approach is applicable to any living organism that can be grown in the field or in culture. The experimental approach is outlined in detail in Knudsen <i>et al</i>., <span>2022</span> and illustrated schematically in Figure 1b. In total, more than 500 000 FIND-IT barley variant plants are today available for screening. FIND-IT populations were also developed in other crops and microorganisms using sodium azide or ethyl methanesulphonate (EMS) as mutagens. Thus FIND-IT has been the key technology used to obtain sweet seeds in white lupin (Mancinotti <i>et al</i>., <span>2023</span>), eliminate the presence of anti-nutritional saponins in quinoa seeds (Trinh <i>et al</i>., <span>2024</span>), improve phosphate bioavailability in the barley grain (Madsen <i>et al</i>., <span>2024</span>), avoid hydroxynitrile glucoside-derived formation of the pro-carcinogen ethyl carbamate in whisky production (Figure S1; Jørgensen <i>et al</i>., <span>2024</span>) and modify the flavour profiles of Saccharomyces species for use in industrial brewing (Stovisek <i>et al</i>., <span>2024</span>).</p><p>The FIND-IT technology pipeline was designed based on knowledge of the mutant load and spectrum obtained in barley using different doses of sodium azide as monitored by whole genome sequencing (Figure 1b; Knudsen <i>et al</i>., <span>2022</span>). The introduced mutations were found to be equally distributed over the seven barley chromosomes with a higher number of mutations observed at increased mutagen doses. Thus, FIND-IT libraries can be generated for different purposes: Medium mutation load (<i>e.g</i>. 1.7 m<span>m</span> sodium azide treatment for an average of 14 770 SNPs per individual plant) for gene-function analyses or low mutation load (<i>e.g</i>. 0.3 m<span>m</span> sodium azide treatment for an average of 5565 SNPs per individual plant) for barley breeding (Knudsen <i>et al</i>., <span>2022</span>). The whole-genome sequencing documents that 15% of the barley sodium azide mutations are transversions and 85% are transitions with a preference for C > T and G > A (Figure 1c). In the RGT Planet barley genome, 1900 Mb are C's and G's. Upon sodium azide mutagenesis using an average dose, 10 000 randomly positioned SNPs are introduced in each single plant of which around 8000 will be C > T and G > A transitions. Using the FIND-IT technology, a library collection of 350 000 plants may now be analysed containing approximately 350 000 × 8000 = 2 800 000 000 randomly distributed C > T and G > A transitions (Figure 1b). Because the entire RGT Planet barley genome only harbours 1900 Mb C and G nucleotides, this means that virtually all putative mutation sites in the plant variant population have been saturated. When the sites are saturated, their status shifts from being random to become available as defined distinct sites in one or more individual plants present in the variant collection. Accordingly, the individual grains of the plant carrying a specific desired SNP in a specific gene may be identified from the many grain pools using a TaqMan™ assay and digital PCR (Figure 1b). You know the mutation is there and just have to FIND-IT.</p><p>Compared with existing cereal TILLING resources for variation breeding and in relation to the natural variation found in the barley pan-genome accession panel, the FIND-IT off-target mutation pressure in breeding libraries is low and can be efficiently reduced by backcrossing to the parent or by direct crosses to elite barley cultivars in commercial breeding programmes. In this context, it is to be noticed that the off-target mutation pressure using FIND-IT would typically be adjusted to be comparable in number of SNPs induced in a single classical backcrossing step between two parents with naturally distinct genomes (Knudsen <i>et al</i>., <span>2022</span>).</p><p>As a proof of principle, we demonstrated the efficiency of the FIND-IT technology pipeline by isolating 100 targeted barley gene knockout lines and two dozen lines with specific amino acid exchanges or miRNA and promoter variants (Figure S2; Knudsen <i>et al</i>., <span>2022</span>). Taking advantage of the fact that FIND-IT is a non-GMO approach method, data were directly verified by growing the barley variants in the field (Knudsen <i>et al</i>., <span>2022</span>), an important requirement to validate new crop traits (Khaipho-Burch <i>et al</i>., <span>2023</span>).</p><p>The current development of high-quality plant genome and pan-genome resources allows breeding strategies to become highly customized. While TILLING-by-sequencing resources are static regarding the chosen variant, and with CRISPR technologies still facing multiple challenges to become field-applicable (Cardi <i>et al</i>., <span>2023</span>), the FIND-IT pipeline stays agile, offers high flexibility and high-throughput, and is breeding compatible today. FIND-IT library resources can be regularly updated with new elite lines, customized for specific use (e.g. winter versus spring crop libraries) while the high sensitivity of ddPCR and sample pooling keeps variant screening highly competitive. When isolated from large elite line libraries with low individual mutation load, original FIND-IT variants can be directly implemented in elite breeding protocols to efficiently lose off-target mutations during yield breeding cycles providing unprecedented fast market rollout of novel traits.</p><p>The authors have not declared a conflict of interest.</p>","PeriodicalId":221,"journal":{"name":"Plant Biotechnology Journal","volume":"22 11","pages":"3051-3053"},"PeriodicalIF":10.5000,"publicationDate":"2024-07-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/pbi.14427","citationCount":"0","resultStr":"{\"title\":\"Just FIND-IT: Harnessing the true power of induced mutagenesis\",\"authors\":\"Christoph Dockter, Søren Knudsen, Magnus Wohlfahrt Rasmussen, Birgitte Skadhauge, Birger Lindberg Møller\",\"doi\":\"10.1111/pbi.14427\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>In nature, genetic variation occurs in every population and results in the evolution of a diversity of new properties, some of which promote the survival of the species. To accelerate nature's evolution based on genetic diversity, plant breeders may induce additional mutations to raise the number of genetic variations increasing the chances to obtain varieties with new desired traits like improved nutritive quality, yields and resilience to biotic and abiotic stress factors. Induced mutagenesis based on chemical mutagens is considered non-GM and has been used in barley (<i>Hordeum vulgare</i>) for decades (Hansson <i>et al</i>., <span>2024</span>). Reverse genetic techniques including TILLING (Targeting Induced Local Lesions in Genomes) screening methodology and more recently TILLING-by-sequencing spinoffs are tools used to identify individual plant variants with the desired valuable genomic alterations. However, these tools are hampered by low mutation capacity.</p><p>TILLING is a PCR-based technique designed to detect mismatched single nucleotides in a target gene. In 2023, Szarejko and her research group in Poland published a thorough overview of the TILLING success stories within the last 20 years (Szurman-Zubrzycka <i>et al</i>., <span>2023</span>) including a description of the TILLING populations in different barley cultivars and landraces obtained following chemical mutagenesis (Figure 1a). The TILLING population sizes range between 1372 and 9600 individual plant variants. The mutation frequencies are individually chosen and dose-dependent (1/154–1/2500 Kbp). When multiplied (# of individuals × mutations per individual), the total number of mutations present in barley TILLING populations ranges between 10 and 100 million (Figure 1a). This may sound like a lot, but with a barley genome size of around 4300 Mbp (here, RGT Planet; Jayakodi <i>et al</i>., <span>2020</span>), less than 2% of the nucleotides in the entire population are mutated. This severely reduces the possibility to find a desired mutation in TILLING populations. The FIND-IT technology is a new approach overriding these constraints.</p><p>The FIND-IT technology was published in Science Advances in 2022 (Figure 1b) (Knudsen <i>et al</i>., <span>2022</span>) and provides an agile and high-throughput approach to screen unprecedented large size chemically induced variant populations. FIND-IT combines systematic sample pooling and splitting with high-sensitivity, droplet digital PCR (ddPCR)–based genotyping for targeted identification of desired traits at single-nucleotide resolution. The ddPCR technology is 1000-fold more sensitive than conventional PCR. The FIND-IT approach is applicable to any living organism that can be grown in the field or in culture. The experimental approach is outlined in detail in Knudsen <i>et al</i>., <span>2022</span> and illustrated schematically in Figure 1b. In total, more than 500 000 FIND-IT barley variant plants are today available for screening. FIND-IT populations were also developed in other crops and microorganisms using sodium azide or ethyl methanesulphonate (EMS) as mutagens. Thus FIND-IT has been the key technology used to obtain sweet seeds in white lupin (Mancinotti <i>et al</i>., <span>2023</span>), eliminate the presence of anti-nutritional saponins in quinoa seeds (Trinh <i>et al</i>., <span>2024</span>), improve phosphate bioavailability in the barley grain (Madsen <i>et al</i>., <span>2024</span>), avoid hydroxynitrile glucoside-derived formation of the pro-carcinogen ethyl carbamate in whisky production (Figure S1; Jørgensen <i>et al</i>., <span>2024</span>) and modify the flavour profiles of Saccharomyces species for use in industrial brewing (Stovisek <i>et al</i>., <span>2024</span>).</p><p>The FIND-IT technology pipeline was designed based on knowledge of the mutant load and spectrum obtained in barley using different doses of sodium azide as monitored by whole genome sequencing (Figure 1b; Knudsen <i>et al</i>., <span>2022</span>). The introduced mutations were found to be equally distributed over the seven barley chromosomes with a higher number of mutations observed at increased mutagen doses. Thus, FIND-IT libraries can be generated for different purposes: Medium mutation load (<i>e.g</i>. 1.7 m<span>m</span> sodium azide treatment for an average of 14 770 SNPs per individual plant) for gene-function analyses or low mutation load (<i>e.g</i>. 0.3 m<span>m</span> sodium azide treatment for an average of 5565 SNPs per individual plant) for barley breeding (Knudsen <i>et al</i>., <span>2022</span>). The whole-genome sequencing documents that 15% of the barley sodium azide mutations are transversions and 85% are transitions with a preference for C > T and G > A (Figure 1c). In the RGT Planet barley genome, 1900 Mb are C's and G's. Upon sodium azide mutagenesis using an average dose, 10 000 randomly positioned SNPs are introduced in each single plant of which around 8000 will be C > T and G > A transitions. Using the FIND-IT technology, a library collection of 350 000 plants may now be analysed containing approximately 350 000 × 8000 = 2 800 000 000 randomly distributed C > T and G > A transitions (Figure 1b). Because the entire RGT Planet barley genome only harbours 1900 Mb C and G nucleotides, this means that virtually all putative mutation sites in the plant variant population have been saturated. When the sites are saturated, their status shifts from being random to become available as defined distinct sites in one or more individual plants present in the variant collection. Accordingly, the individual grains of the plant carrying a specific desired SNP in a specific gene may be identified from the many grain pools using a TaqMan™ assay and digital PCR (Figure 1b). You know the mutation is there and just have to FIND-IT.</p><p>Compared with existing cereal TILLING resources for variation breeding and in relation to the natural variation found in the barley pan-genome accession panel, the FIND-IT off-target mutation pressure in breeding libraries is low and can be efficiently reduced by backcrossing to the parent or by direct crosses to elite barley cultivars in commercial breeding programmes. In this context, it is to be noticed that the off-target mutation pressure using FIND-IT would typically be adjusted to be comparable in number of SNPs induced in a single classical backcrossing step between two parents with naturally distinct genomes (Knudsen <i>et al</i>., <span>2022</span>).</p><p>As a proof of principle, we demonstrated the efficiency of the FIND-IT technology pipeline by isolating 100 targeted barley gene knockout lines and two dozen lines with specific amino acid exchanges or miRNA and promoter variants (Figure S2; Knudsen <i>et al</i>., <span>2022</span>). Taking advantage of the fact that FIND-IT is a non-GMO approach method, data were directly verified by growing the barley variants in the field (Knudsen <i>et al</i>., <span>2022</span>), an important requirement to validate new crop traits (Khaipho-Burch <i>et al</i>., <span>2023</span>).</p><p>The current development of high-quality plant genome and pan-genome resources allows breeding strategies to become highly customized. While TILLING-by-sequencing resources are static regarding the chosen variant, and with CRISPR technologies still facing multiple challenges to become field-applicable (Cardi <i>et al</i>., <span>2023</span>), the FIND-IT pipeline stays agile, offers high flexibility and high-throughput, and is breeding compatible today. FIND-IT library resources can be regularly updated with new elite lines, customized for specific use (e.g. winter versus spring crop libraries) while the high sensitivity of ddPCR and sample pooling keeps variant screening highly competitive. When isolated from large elite line libraries with low individual mutation load, original FIND-IT variants can be directly implemented in elite breeding protocols to efficiently lose off-target mutations during yield breeding cycles providing unprecedented fast market rollout of novel traits.</p><p>The authors have not declared a conflict of interest.</p>\",\"PeriodicalId\":221,\"journal\":{\"name\":\"Plant Biotechnology Journal\",\"volume\":\"22 11\",\"pages\":\"3051-3053\"},\"PeriodicalIF\":10.5000,\"publicationDate\":\"2024-07-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/pbi.14427\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Plant Biotechnology Journal\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/pbi.14427\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Plant Biotechnology Journal","FirstCategoryId":"5","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/pbi.14427","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
Just FIND-IT: Harnessing the true power of induced mutagenesis
In nature, genetic variation occurs in every population and results in the evolution of a diversity of new properties, some of which promote the survival of the species. To accelerate nature's evolution based on genetic diversity, plant breeders may induce additional mutations to raise the number of genetic variations increasing the chances to obtain varieties with new desired traits like improved nutritive quality, yields and resilience to biotic and abiotic stress factors. Induced mutagenesis based on chemical mutagens is considered non-GM and has been used in barley (Hordeum vulgare) for decades (Hansson et al., 2024). Reverse genetic techniques including TILLING (Targeting Induced Local Lesions in Genomes) screening methodology and more recently TILLING-by-sequencing spinoffs are tools used to identify individual plant variants with the desired valuable genomic alterations. However, these tools are hampered by low mutation capacity.
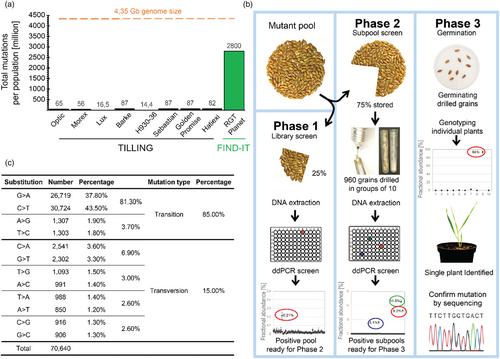
TILLING is a PCR-based technique designed to detect mismatched single nucleotides in a target gene. In 2023, Szarejko and her research group in Poland published a thorough overview of the TILLING success stories within the last 20 years (Szurman-Zubrzycka et al., 2023) including a description of the TILLING populations in different barley cultivars and landraces obtained following chemical mutagenesis (Figure 1a). The TILLING population sizes range between 1372 and 9600 individual plant variants. The mutation frequencies are individually chosen and dose-dependent (1/154–1/2500 Kbp). When multiplied (# of individuals × mutations per individual), the total number of mutations present in barley TILLING populations ranges between 10 and 100 million (Figure 1a). This may sound like a lot, but with a barley genome size of around 4300 Mbp (here, RGT Planet; Jayakodi et al., 2020), less than 2% of the nucleotides in the entire population are mutated. This severely reduces the possibility to find a desired mutation in TILLING populations. The FIND-IT technology is a new approach overriding these constraints.
The FIND-IT technology was published in Science Advances in 2022 (Figure 1b) (Knudsen et al., 2022) and provides an agile and high-throughput approach to screen unprecedented large size chemically induced variant populations. FIND-IT combines systematic sample pooling and splitting with high-sensitivity, droplet digital PCR (ddPCR)–based genotyping for targeted identification of desired traits at single-nucleotide resolution. The ddPCR technology is 1000-fold more sensitive than conventional PCR. The FIND-IT approach is applicable to any living organism that can be grown in the field or in culture. The experimental approach is outlined in detail in Knudsen et al., 2022 and illustrated schematically in Figure 1b. In total, more than 500 000 FIND-IT barley variant plants are today available for screening. FIND-IT populations were also developed in other crops and microorganisms using sodium azide or ethyl methanesulphonate (EMS) as mutagens. Thus FIND-IT has been the key technology used to obtain sweet seeds in white lupin (Mancinotti et al., 2023), eliminate the presence of anti-nutritional saponins in quinoa seeds (Trinh et al., 2024), improve phosphate bioavailability in the barley grain (Madsen et al., 2024), avoid hydroxynitrile glucoside-derived formation of the pro-carcinogen ethyl carbamate in whisky production (Figure S1; Jørgensen et al., 2024) and modify the flavour profiles of Saccharomyces species for use in industrial brewing (Stovisek et al., 2024).
The FIND-IT technology pipeline was designed based on knowledge of the mutant load and spectrum obtained in barley using different doses of sodium azide as monitored by whole genome sequencing (Figure 1b; Knudsen et al., 2022). The introduced mutations were found to be equally distributed over the seven barley chromosomes with a higher number of mutations observed at increased mutagen doses. Thus, FIND-IT libraries can be generated for different purposes: Medium mutation load (e.g. 1.7 mm sodium azide treatment for an average of 14 770 SNPs per individual plant) for gene-function analyses or low mutation load (e.g. 0.3 mm sodium azide treatment for an average of 5565 SNPs per individual plant) for barley breeding (Knudsen et al., 2022). The whole-genome sequencing documents that 15% of the barley sodium azide mutations are transversions and 85% are transitions with a preference for C > T and G > A (Figure 1c). In the RGT Planet barley genome, 1900 Mb are C's and G's. Upon sodium azide mutagenesis using an average dose, 10 000 randomly positioned SNPs are introduced in each single plant of which around 8000 will be C > T and G > A transitions. Using the FIND-IT technology, a library collection of 350 000 plants may now be analysed containing approximately 350 000 × 8000 = 2 800 000 000 randomly distributed C > T and G > A transitions (Figure 1b). Because the entire RGT Planet barley genome only harbours 1900 Mb C and G nucleotides, this means that virtually all putative mutation sites in the plant variant population have been saturated. When the sites are saturated, their status shifts from being random to become available as defined distinct sites in one or more individual plants present in the variant collection. Accordingly, the individual grains of the plant carrying a specific desired SNP in a specific gene may be identified from the many grain pools using a TaqMan™ assay and digital PCR (Figure 1b). You know the mutation is there and just have to FIND-IT.
Compared with existing cereal TILLING resources for variation breeding and in relation to the natural variation found in the barley pan-genome accession panel, the FIND-IT off-target mutation pressure in breeding libraries is low and can be efficiently reduced by backcrossing to the parent or by direct crosses to elite barley cultivars in commercial breeding programmes. In this context, it is to be noticed that the off-target mutation pressure using FIND-IT would typically be adjusted to be comparable in number of SNPs induced in a single classical backcrossing step between two parents with naturally distinct genomes (Knudsen et al., 2022).
As a proof of principle, we demonstrated the efficiency of the FIND-IT technology pipeline by isolating 100 targeted barley gene knockout lines and two dozen lines with specific amino acid exchanges or miRNA and promoter variants (Figure S2; Knudsen et al., 2022). Taking advantage of the fact that FIND-IT is a non-GMO approach method, data were directly verified by growing the barley variants in the field (Knudsen et al., 2022), an important requirement to validate new crop traits (Khaipho-Burch et al., 2023).
The current development of high-quality plant genome and pan-genome resources allows breeding strategies to become highly customized. While TILLING-by-sequencing resources are static regarding the chosen variant, and with CRISPR technologies still facing multiple challenges to become field-applicable (Cardi et al., 2023), the FIND-IT pipeline stays agile, offers high flexibility and high-throughput, and is breeding compatible today. FIND-IT library resources can be regularly updated with new elite lines, customized for specific use (e.g. winter versus spring crop libraries) while the high sensitivity of ddPCR and sample pooling keeps variant screening highly competitive. When isolated from large elite line libraries with low individual mutation load, original FIND-IT variants can be directly implemented in elite breeding protocols to efficiently lose off-target mutations during yield breeding cycles providing unprecedented fast market rollout of novel traits.
The authors have not declared a conflict of interest.
期刊介绍:
Plant Biotechnology Journal aspires to publish original research and insightful reviews of high impact, authored by prominent researchers in applied plant science. The journal places a special emphasis on molecular plant sciences and their practical applications through plant biotechnology. Our goal is to establish a platform for showcasing significant advances in the field, encompassing curiosity-driven studies with potential applications, strategic research in plant biotechnology, scientific analysis of crucial issues for the beneficial utilization of plant sciences, and assessments of the performance of plant biotechnology products in practical applications.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
