Ashley Hirons, David Yurick, Natasha Jansz, Paula Ellenberg, Genoveffa Franchini, Lloyd Einsiedel, Georges Khoury, Damian F J Purcell
{"title":"澳大利亚中部地区 HTLV-1 亚型-C 的 orf-I p12 和 hbz 基因存在高度基因组差异。","authors":"Ashley Hirons, David Yurick, Natasha Jansz, Paula Ellenberg, Genoveffa Franchini, Lloyd Einsiedel, Georges Khoury, Damian F J Purcell","doi":"10.1186/s12977-024-00647-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Human T cell lymphotropic virus type 1 (HTLV-1) infection remains a largely neglected public health problem, particularly in resource-poor areas with high burden of communicable and non-communicable diseases, such as some remote populations in Central Australia where an estimated 37% of adults are infected with HTLV-1. Most of our understanding of HTLV-1 infection comes from studies of the globally spread subtype-A (HTLV-1a), with few molecular studies reported with the Austral-Melanesian subtype-C (HTLV-1c) predominant in the Indo-Pacific and Oceania regions.</p><p><strong>Results: </strong>Using a primer walking strategy and direct sequencing, we constructed HTLV-1c genomic consensus sequences from 22 First Nations participants living with HTLV-1c in Central Australia. Phylogenetic and pairwise analysis of this subtype-C proviral gDNA showed higher levels of genomic divergence in comparison to previously published HTLV-1a genomes. While the overall genomic homology between subtypes was 92.5%, the lowest nucleotide and amino acid sequence identity occurred near the 3' end of the proviral genome coding regulatory genes, especially overlapping hbz (85.37%, 77.46%, respectively) and orf-I product p12 (82.00%, 70.30%, respectively). Strikingly, the HTLV-1c genomic consensus sequences uniformly showed a defective translation start codon for the immune regulatory proteins p12/p8 encoded by the HTLV-1A orf-I. Deletions in the proviral genome were detected in many subjects, particularly in the structural gag, pol and env genes. Similarly, using a droplet digital PCR assay measuring the copies of gag and tax per reference host genome, we quantitatively confirmed that provirus retains the tax gene region at higher levels than gag.</p><p><strong>Conclusions: </strong>Our genomic analysis of HTLV-1c in Central Australia in conjunction with earlier Melanesian HTLV-1c sequences, elucidate substantial differences with respect to the globally spread HTLV-1a. Future studies should address the impact these genomic differences have on infection and the regionally distinctive frequency of associated pulmonary disease. Understanding the host and virus subtype factors which contribute to the differential morbidity observed, is crucial for the development of much needed therapeutics and vaccine strategies against this highly endemic infection in remote First Nations communities in Central Australia.</p>","PeriodicalId":21123,"journal":{"name":"Retrovirology","volume":"21 1","pages":"14"},"PeriodicalIF":4.1000,"publicationDate":"2024-07-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11253349/pdf/","citationCount":"0","resultStr":"{\"title\":\"High level of genomic divergence in orf-I p12 and hbz genes of HTLV-1 subtype-C in Central Australia.\",\"authors\":\"Ashley Hirons, David Yurick, Natasha Jansz, Paula Ellenberg, Genoveffa Franchini, Lloyd Einsiedel, Georges Khoury, Damian F J Purcell\",\"doi\":\"10.1186/s12977-024-00647-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Human T cell lymphotropic virus type 1 (HTLV-1) infection remains a largely neglected public health problem, particularly in resource-poor areas with high burden of communicable and non-communicable diseases, such as some remote populations in Central Australia where an estimated 37% of adults are infected with HTLV-1. Most of our understanding of HTLV-1 infection comes from studies of the globally spread subtype-A (HTLV-1a), with few molecular studies reported with the Austral-Melanesian subtype-C (HTLV-1c) predominant in the Indo-Pacific and Oceania regions.</p><p><strong>Results: </strong>Using a primer walking strategy and direct sequencing, we constructed HTLV-1c genomic consensus sequences from 22 First Nations participants living with HTLV-1c in Central Australia. Phylogenetic and pairwise analysis of this subtype-C proviral gDNA showed higher levels of genomic divergence in comparison to previously published HTLV-1a genomes. While the overall genomic homology between subtypes was 92.5%, the lowest nucleotide and amino acid sequence identity occurred near the 3' end of the proviral genome coding regulatory genes, especially overlapping hbz (85.37%, 77.46%, respectively) and orf-I product p12 (82.00%, 70.30%, respectively). Strikingly, the HTLV-1c genomic consensus sequences uniformly showed a defective translation start codon for the immune regulatory proteins p12/p8 encoded by the HTLV-1A orf-I. Deletions in the proviral genome were detected in many subjects, particularly in the structural gag, pol and env genes. Similarly, using a droplet digital PCR assay measuring the copies of gag and tax per reference host genome, we quantitatively confirmed that provirus retains the tax gene region at higher levels than gag.</p><p><strong>Conclusions: </strong>Our genomic analysis of HTLV-1c in Central Australia in conjunction with earlier Melanesian HTLV-1c sequences, elucidate substantial differences with respect to the globally spread HTLV-1a. Future studies should address the impact these genomic differences have on infection and the regionally distinctive frequency of associated pulmonary disease. Understanding the host and virus subtype factors which contribute to the differential morbidity observed, is crucial for the development of much needed therapeutics and vaccine strategies against this highly endemic infection in remote First Nations communities in Central Australia.</p>\",\"PeriodicalId\":21123,\"journal\":{\"name\":\"Retrovirology\",\"volume\":\"21 1\",\"pages\":\"14\"},\"PeriodicalIF\":4.1000,\"publicationDate\":\"2024-07-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11253349/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Retrovirology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12977-024-00647-w\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"VIROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Retrovirology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12977-024-00647-w","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"VIROLOGY","Score":null,"Total":0}
High level of genomic divergence in orf-I p12 and hbz genes of HTLV-1 subtype-C in Central Australia.
Background: Human T cell lymphotropic virus type 1 (HTLV-1) infection remains a largely neglected public health problem, particularly in resource-poor areas with high burden of communicable and non-communicable diseases, such as some remote populations in Central Australia where an estimated 37% of adults are infected with HTLV-1. Most of our understanding of HTLV-1 infection comes from studies of the globally spread subtype-A (HTLV-1a), with few molecular studies reported with the Austral-Melanesian subtype-C (HTLV-1c) predominant in the Indo-Pacific and Oceania regions.
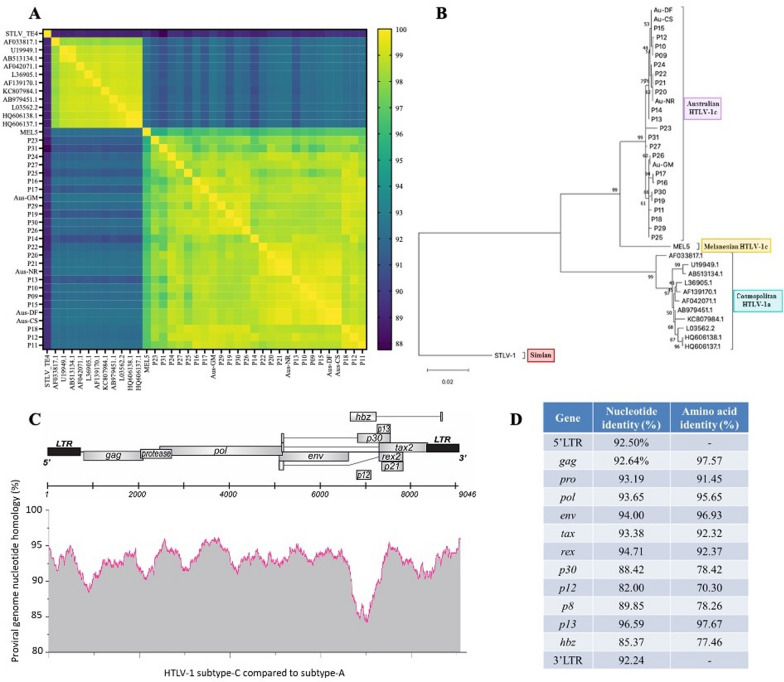
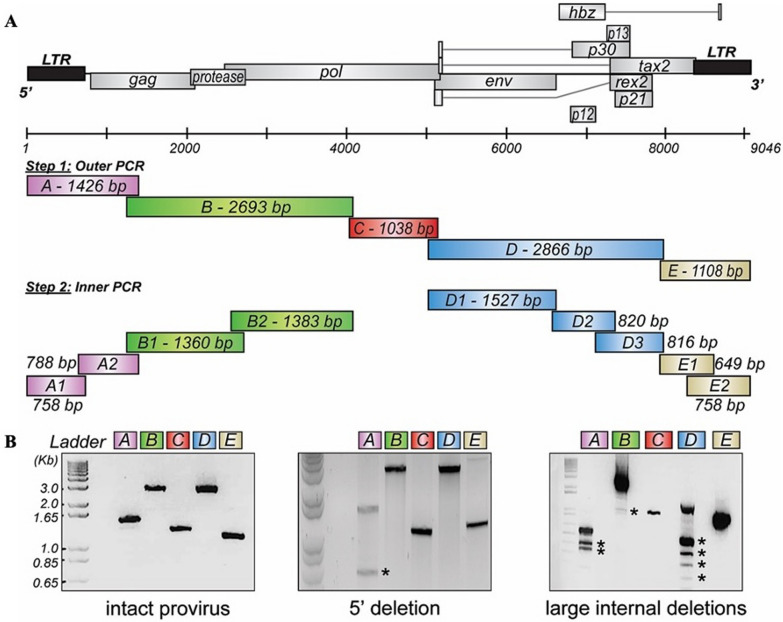
Results: Using a primer walking strategy and direct sequencing, we constructed HTLV-1c genomic consensus sequences from 22 First Nations participants living with HTLV-1c in Central Australia. Phylogenetic and pairwise analysis of this subtype-C proviral gDNA showed higher levels of genomic divergence in comparison to previously published HTLV-1a genomes. While the overall genomic homology between subtypes was 92.5%, the lowest nucleotide and amino acid sequence identity occurred near the 3' end of the proviral genome coding regulatory genes, especially overlapping hbz (85.37%, 77.46%, respectively) and orf-I product p12 (82.00%, 70.30%, respectively). Strikingly, the HTLV-1c genomic consensus sequences uniformly showed a defective translation start codon for the immune regulatory proteins p12/p8 encoded by the HTLV-1A orf-I. Deletions in the proviral genome were detected in many subjects, particularly in the structural gag, pol and env genes. Similarly, using a droplet digital PCR assay measuring the copies of gag and tax per reference host genome, we quantitatively confirmed that provirus retains the tax gene region at higher levels than gag.
Conclusions: Our genomic analysis of HTLV-1c in Central Australia in conjunction with earlier Melanesian HTLV-1c sequences, elucidate substantial differences with respect to the globally spread HTLV-1a. Future studies should address the impact these genomic differences have on infection and the regionally distinctive frequency of associated pulmonary disease. Understanding the host and virus subtype factors which contribute to the differential morbidity observed, is crucial for the development of much needed therapeutics and vaccine strategies against this highly endemic infection in remote First Nations communities in Central Australia.
期刊介绍:
Retrovirology is an open access, online journal that publishes stringently peer-reviewed, high-impact articles on host-pathogen interactions, fundamental mechanisms of replication, immune defenses, animal models, and clinical science relating to retroviruses. Retroviruses are pleiotropically found in animals. Well-described examples include avian, murine and primate retroviruses.
Two human retroviruses are especially important pathogens. These are the human immunodeficiency virus, HIV, and the human T-cell leukemia virus, HTLV. HIV causes AIDS while HTLV-1 is the etiological agent for adult T-cell leukemia and HTLV-1-associated myelopathy/tropical spastic paraparesis. Retrovirology aims to cover comprehensively all aspects of human and animal retrovirus research.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
