Hemchandra Deka, Atul Pawar, Monishka Battula, Ayman A. Ghfar, Mohamed E. Assal, Rupesh V. Chikhale
{"title":"针对分枝杆菌蛋白激酶 PknB 的新型潜在抗菌肽的鉴定和设计","authors":"Hemchandra Deka, Atul Pawar, Monishka Battula, Ayman A. Ghfar, Mohamed E. Assal, Rupesh V. Chikhale","doi":"10.1007/s10930-024-10218-9","DOIUrl":null,"url":null,"abstract":"<div><p>Antimicrobial peptides have gradually gained advantages over small molecule inhibitors for their multifunctional effects, synthesising accessibility and target specificity. The current study aims to determine an antimicrobial peptide to inhibit PknB, a serine/threonine protein kinase (STPK), by binding efficiently at the helically oriented hinge region. A library of 5626 antimicrobial peptides from publicly available repositories has been prepared and categorised based on the length. Molecular docking using ADCP helped to find the multiple conformations of the subjected peptides. For each peptide served as input the tool outputs 100 poses of the subjected peptide. To maintain an efficient binding for relatively a longer duration, only those peptides were chosen which were seen to bind constantly to the active site of the receptor protein over all the poses observed. Each peptide had different number of constituent amino acid residues; the peptides were classified based on the length into five groups. In each group the peptide length incremented upto four residues from the initial length form. Five peptides were selected for Molecular Dynamic simulation in Gromacs based on higher binding affinity. Post-dynamic analysis and the frame comparison inferred that neither the shorter nor the longer peptide but an intermediate length of 15 mer peptide bound well to the receptor. Residual substitution to the selected peptides was performed to enhance the targeted interaction. The new complexes considered were further analysed using the Elastic Network Model (ENM) for the functional site’s intrinsic dynamic movement to estimate the new peptide’s role. The study sheds light on prospects that besides the length of peptides, the combination of constituent residues equally plays a pivotal role in peptide-based inhibitor generation. The study envisages the challenges of fine-tuned peptide recovery and the scope of Machine Learning (ML) and Deep Learning (DL) algorithm development. As the study was primarily meant for generation of therapeutics for Tuberculosis (TB), the peptide proposed by this study demands meticulous invitro analysis prior to clinical applications.</p><h3>Graphical Abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":793,"journal":{"name":"The Protein Journal","volume":"43 4","pages":"858 - 868"},"PeriodicalIF":1.4000,"publicationDate":"2024-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11345320/pdf/","citationCount":"0","resultStr":"{\"title\":\"Identification and Design of Novel Potential Antimicrobial Peptides Targeting Mycobacterial Protein Kinase PknB\",\"authors\":\"Hemchandra Deka, Atul Pawar, Monishka Battula, Ayman A. Ghfar, Mohamed E. Assal, Rupesh V. Chikhale\",\"doi\":\"10.1007/s10930-024-10218-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Antimicrobial peptides have gradually gained advantages over small molecule inhibitors for their multifunctional effects, synthesising accessibility and target specificity. The current study aims to determine an antimicrobial peptide to inhibit PknB, a serine/threonine protein kinase (STPK), by binding efficiently at the helically oriented hinge region. A library of 5626 antimicrobial peptides from publicly available repositories has been prepared and categorised based on the length. Molecular docking using ADCP helped to find the multiple conformations of the subjected peptides. For each peptide served as input the tool outputs 100 poses of the subjected peptide. To maintain an efficient binding for relatively a longer duration, only those peptides were chosen which were seen to bind constantly to the active site of the receptor protein over all the poses observed. Each peptide had different number of constituent amino acid residues; the peptides were classified based on the length into five groups. In each group the peptide length incremented upto four residues from the initial length form. Five peptides were selected for Molecular Dynamic simulation in Gromacs based on higher binding affinity. Post-dynamic analysis and the frame comparison inferred that neither the shorter nor the longer peptide but an intermediate length of 15 mer peptide bound well to the receptor. Residual substitution to the selected peptides was performed to enhance the targeted interaction. The new complexes considered were further analysed using the Elastic Network Model (ENM) for the functional site’s intrinsic dynamic movement to estimate the new peptide’s role. The study sheds light on prospects that besides the length of peptides, the combination of constituent residues equally plays a pivotal role in peptide-based inhibitor generation. The study envisages the challenges of fine-tuned peptide recovery and the scope of Machine Learning (ML) and Deep Learning (DL) algorithm development. As the study was primarily meant for generation of therapeutics for Tuberculosis (TB), the peptide proposed by this study demands meticulous invitro analysis prior to clinical applications.</p><h3>Graphical Abstract</h3>\\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>\",\"PeriodicalId\":793,\"journal\":{\"name\":\"The Protein Journal\",\"volume\":\"43 4\",\"pages\":\"858 - 868\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2024-07-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11345320/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Protein Journal\",\"FirstCategoryId\":\"2\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10930-024-10218-9\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Protein Journal","FirstCategoryId":"2","ListUrlMain":"https://link.springer.com/article/10.1007/s10930-024-10218-9","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Identification and Design of Novel Potential Antimicrobial Peptides Targeting Mycobacterial Protein Kinase PknB
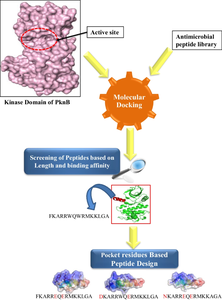
Antimicrobial peptides have gradually gained advantages over small molecule inhibitors for their multifunctional effects, synthesising accessibility and target specificity. The current study aims to determine an antimicrobial peptide to inhibit PknB, a serine/threonine protein kinase (STPK), by binding efficiently at the helically oriented hinge region. A library of 5626 antimicrobial peptides from publicly available repositories has been prepared and categorised based on the length. Molecular docking using ADCP helped to find the multiple conformations of the subjected peptides. For each peptide served as input the tool outputs 100 poses of the subjected peptide. To maintain an efficient binding for relatively a longer duration, only those peptides were chosen which were seen to bind constantly to the active site of the receptor protein over all the poses observed. Each peptide had different number of constituent amino acid residues; the peptides were classified based on the length into five groups. In each group the peptide length incremented upto four residues from the initial length form. Five peptides were selected for Molecular Dynamic simulation in Gromacs based on higher binding affinity. Post-dynamic analysis and the frame comparison inferred that neither the shorter nor the longer peptide but an intermediate length of 15 mer peptide bound well to the receptor. Residual substitution to the selected peptides was performed to enhance the targeted interaction. The new complexes considered were further analysed using the Elastic Network Model (ENM) for the functional site’s intrinsic dynamic movement to estimate the new peptide’s role. The study sheds light on prospects that besides the length of peptides, the combination of constituent residues equally plays a pivotal role in peptide-based inhibitor generation. The study envisages the challenges of fine-tuned peptide recovery and the scope of Machine Learning (ML) and Deep Learning (DL) algorithm development. As the study was primarily meant for generation of therapeutics for Tuberculosis (TB), the peptide proposed by this study demands meticulous invitro analysis prior to clinical applications.
期刊介绍:
The Protein Journal (formerly the Journal of Protein Chemistry) publishes original research work on all aspects of proteins and peptides. These include studies concerned with covalent or three-dimensional structure determination (X-ray, NMR, cryoEM, EPR/ESR, optical methods, etc.), computational aspects of protein structure and function, protein folding and misfolding, assembly, genetics, evolution, proteomics, molecular biology, protein engineering, protein nanotechnology, protein purification and analysis and peptide synthesis, as well as the elucidation and interpretation of the molecular bases of biological activities of proteins and peptides. We accept original research papers, reviews, mini-reviews, hypotheses, opinion papers, and letters to the editor.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
