{"title":"利用机器学习揭示 IIb 类氨基酰-tRNA 合成酶的底物特异性决定因素","authors":"Thomas Simonson, Victor Mihaila, Ivan Reveguk","doi":"10.1016/j.jmgm.2024.108818","DOIUrl":null,"url":null,"abstract":"<div><p>Specific amino acid (AA) binding by aminoacyl-tRNA synthetases (aaRSs) is necessary for correct translation of the genetic code. Sequence and structure analyses have revealed the main specificity determinants and allowed a partitioning of aaRSs into two classes and several subclasses. However, the information contributed by each determinant has not been precisely quantified, and other, minor determinants may still be unidentified. Growth of genomic data and development of machine learning classification methods allow us to revisit these questions. This work considered the subclass IIb, formed by the three enzymes aspartyl-, asparaginyl-, and lysyl-tRNA synthetase (LysRS). Over 35,000 sequences from the Pfam database were considered, and used to train a machine-learning model based on ensembles of decision trees. The model was trained to reproduce the existing classification of each sequence as AspRS, AsnRS, or LysRS, and to identify which sequence positions were most important for the classification. A few positions (5–8 depending on the AA substrate) sufficed for accurate classification. Most but not all of them were well-known specificity determinants. The machine learning models thus identified sets of mutations that distinguish the three subclass members, which might be targeted in engineering efforts to alter or swap the AA specificities for biotechnology applications.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"132 ","pages":"Article 108818"},"PeriodicalIF":3.1000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Uncovering substrate specificity determinants of class IIb aminoacyl-tRNA synthetases with machine learning\",\"authors\":\"Thomas Simonson, Victor Mihaila, Ivan Reveguk\",\"doi\":\"10.1016/j.jmgm.2024.108818\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Specific amino acid (AA) binding by aminoacyl-tRNA synthetases (aaRSs) is necessary for correct translation of the genetic code. Sequence and structure analyses have revealed the main specificity determinants and allowed a partitioning of aaRSs into two classes and several subclasses. However, the information contributed by each determinant has not been precisely quantified, and other, minor determinants may still be unidentified. Growth of genomic data and development of machine learning classification methods allow us to revisit these questions. This work considered the subclass IIb, formed by the three enzymes aspartyl-, asparaginyl-, and lysyl-tRNA synthetase (LysRS). Over 35,000 sequences from the Pfam database were considered, and used to train a machine-learning model based on ensembles of decision trees. The model was trained to reproduce the existing classification of each sequence as AspRS, AsnRS, or LysRS, and to identify which sequence positions were most important for the classification. A few positions (5–8 depending on the AA substrate) sufficed for accurate classification. Most but not all of them were well-known specificity determinants. The machine learning models thus identified sets of mutations that distinguish the three subclass members, which might be targeted in engineering efforts to alter or swap the AA specificities for biotechnology applications.</p></div>\",\"PeriodicalId\":16361,\"journal\":{\"name\":\"Journal of molecular graphics & modelling\",\"volume\":\"132 \",\"pages\":\"Article 108818\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular graphics & modelling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1093326324001189\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/7/14 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001189","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/14 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
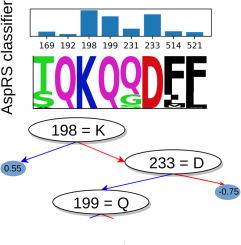
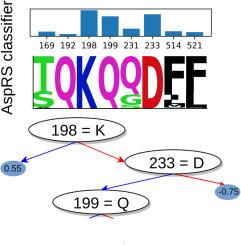
氨基酰-tRNA 合成酶(aaRSs)的特异性氨基酸(AA)结合是正确翻译遗传密码的必要条件。序列和结构分析揭示了主要的特异性决定因素,并将 aaRS 分成两类和若干亚类。然而,每个决定因素所贡献的信息尚未精确量化,其他次要决定因素可能仍未确定。基因组数据的增长和机器学习分类方法的发展使我们能够重新审视这些问题。这项工作研究了由天冬氨酰、天冬氨酰和赖氨酰-tRNA 合成酶(LysRS)三种酶组成的 IIb 亚类。研究考虑了 Pfam 数据库中的 35,000 多个序列,并利用这些序列训练了一个基于决策树集合的机器学习模型。训练该模型的目的是重现每个序列作为 AspRS、AsnRS 或 LysRS 的现有分类,并确定哪些序列位置对分类最重要。有几个位置(5-8 个,取决于 AA 底物)足以进行准确分类。其中大部分(但不是全部)是众所周知的特异性决定因素。因此,机器学习模型确定了区分三个亚类成员的突变集,这些突变集可能是改变或交换 AA 特异性的生物技术应用工程中的目标。
Uncovering substrate specificity determinants of class IIb aminoacyl-tRNA synthetases with machine learning
Specific amino acid (AA) binding by aminoacyl-tRNA synthetases (aaRSs) is necessary for correct translation of the genetic code. Sequence and structure analyses have revealed the main specificity determinants and allowed a partitioning of aaRSs into two classes and several subclasses. However, the information contributed by each determinant has not been precisely quantified, and other, minor determinants may still be unidentified. Growth of genomic data and development of machine learning classification methods allow us to revisit these questions. This work considered the subclass IIb, formed by the three enzymes aspartyl-, asparaginyl-, and lysyl-tRNA synthetase (LysRS). Over 35,000 sequences from the Pfam database were considered, and used to train a machine-learning model based on ensembles of decision trees. The model was trained to reproduce the existing classification of each sequence as AspRS, AsnRS, or LysRS, and to identify which sequence positions were most important for the classification. A few positions (5–8 depending on the AA substrate) sufficed for accurate classification. Most but not all of them were well-known specificity determinants. The machine learning models thus identified sets of mutations that distinguish the three subclass members, which might be targeted in engineering efforts to alter or swap the AA specificities for biotechnology applications.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
