Ben Pode-Shakked, Yuval E. Landau, Nava Shaul Lotan, Joshua Manor, Nitsan Haham, Eyal Kristal, Eli Hershkovitz, Guy Hazan, Yarden Haham, Shlomo Almashanu, Yair Anikster, Orna Staretz-Chacham
{"title":"以色列二氢脂酰胺脱氢酶缺乏症的自然史。","authors":"Ben Pode-Shakked, Yuval E. Landau, Nava Shaul Lotan, Joshua Manor, Nitsan Haham, Eyal Kristal, Eli Hershkovitz, Guy Hazan, Yarden Haham, Shlomo Almashanu, Yair Anikster, Orna Staretz-Chacham","doi":"10.1002/jimd.12778","DOIUrl":null,"url":null,"abstract":"<p>Dihydrolipoamide dehydrogenase (DLD) deficiency is an ultra-rare autosomal-recessive inborn error of metabolism, affecting no less than five mitochondrial multienzyme complexes. With approximately 30 patients reported to date, DLD deficiency was associated with three major clinical presentations: an early-onset encephalopathic phenotype with metabolic acidosis, a predominantly hepatic presentation with liver failure, and a rare myopathic phenotype. To elucidate the demographic, phenotypic, and molecular characteristics of patients with DLD deficiency within the Israeli population, data were collected from metabolic disease specialists in four large tertiary medical centers in the center and south of Israel. Pediatric and adult patients with biallelic variants in <i>DLD</i> were included in the study. A total of 53 patients of 35 families were included in the cohort. Age at presentation ranged between birth and 10 years. Wide phenotypic variability was observed, from asymptomatic individuals in their sixth decade of life, to severe, neonatal-onset disease with devastating neurological sequelae. Six <i>DLD</i> variants were noted, the most common of which was the c.685G>T (p.G229C) variant in homozygous form (24/53 patients, 45.3%; 13/35 families), observed mostly among patients of Ashkenazi-Jewish descent, followed by the homozygous c.1436A>T (p.D479V) variant, found in 20 patients of Bedouin descent (37.7%; 16/35 families). Overall, patients did not necessarily present as one of the previously described distinct clinical phenotypes. DLD deficiency is a panethnic disorder, with significant phenotypic variability, and comprises a continuum, rather than three distinct clinical presentations.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 5","pages":"895-902"},"PeriodicalIF":3.8000,"publicationDate":"2024-07-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12778","citationCount":"0","resultStr":"{\"title\":\"The natural history of dihydrolipoamide dehydrogenase deficiency in Israel\",\"authors\":\"Ben Pode-Shakked, Yuval E. Landau, Nava Shaul Lotan, Joshua Manor, Nitsan Haham, Eyal Kristal, Eli Hershkovitz, Guy Hazan, Yarden Haham, Shlomo Almashanu, Yair Anikster, Orna Staretz-Chacham\",\"doi\":\"10.1002/jimd.12778\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Dihydrolipoamide dehydrogenase (DLD) deficiency is an ultra-rare autosomal-recessive inborn error of metabolism, affecting no less than five mitochondrial multienzyme complexes. With approximately 30 patients reported to date, DLD deficiency was associated with three major clinical presentations: an early-onset encephalopathic phenotype with metabolic acidosis, a predominantly hepatic presentation with liver failure, and a rare myopathic phenotype. To elucidate the demographic, phenotypic, and molecular characteristics of patients with DLD deficiency within the Israeli population, data were collected from metabolic disease specialists in four large tertiary medical centers in the center and south of Israel. Pediatric and adult patients with biallelic variants in <i>DLD</i> were included in the study. A total of 53 patients of 35 families were included in the cohort. Age at presentation ranged between birth and 10 years. Wide phenotypic variability was observed, from asymptomatic individuals in their sixth decade of life, to severe, neonatal-onset disease with devastating neurological sequelae. Six <i>DLD</i> variants were noted, the most common of which was the c.685G>T (p.G229C) variant in homozygous form (24/53 patients, 45.3%; 13/35 families), observed mostly among patients of Ashkenazi-Jewish descent, followed by the homozygous c.1436A>T (p.D479V) variant, found in 20 patients of Bedouin descent (37.7%; 16/35 families). Overall, patients did not necessarily present as one of the previously described distinct clinical phenotypes. DLD deficiency is a panethnic disorder, with significant phenotypic variability, and comprises a continuum, rather than three distinct clinical presentations.</p>\",\"PeriodicalId\":16281,\"journal\":{\"name\":\"Journal of Inherited Metabolic Disease\",\"volume\":\"47 5\",\"pages\":\"895-902\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2024-07-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12778\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Inherited Metabolic Disease\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12778\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12778","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
The natural history of dihydrolipoamide dehydrogenase deficiency in Israel
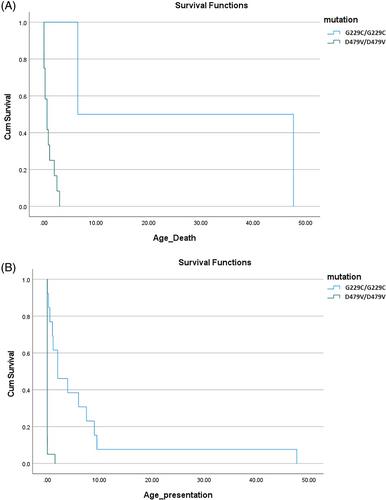
Dihydrolipoamide dehydrogenase (DLD) deficiency is an ultra-rare autosomal-recessive inborn error of metabolism, affecting no less than five mitochondrial multienzyme complexes. With approximately 30 patients reported to date, DLD deficiency was associated with three major clinical presentations: an early-onset encephalopathic phenotype with metabolic acidosis, a predominantly hepatic presentation with liver failure, and a rare myopathic phenotype. To elucidate the demographic, phenotypic, and molecular characteristics of patients with DLD deficiency within the Israeli population, data were collected from metabolic disease specialists in four large tertiary medical centers in the center and south of Israel. Pediatric and adult patients with biallelic variants in DLD were included in the study. A total of 53 patients of 35 families were included in the cohort. Age at presentation ranged between birth and 10 years. Wide phenotypic variability was observed, from asymptomatic individuals in their sixth decade of life, to severe, neonatal-onset disease with devastating neurological sequelae. Six DLD variants were noted, the most common of which was the c.685G>T (p.G229C) variant in homozygous form (24/53 patients, 45.3%; 13/35 families), observed mostly among patients of Ashkenazi-Jewish descent, followed by the homozygous c.1436A>T (p.D479V) variant, found in 20 patients of Bedouin descent (37.7%; 16/35 families). Overall, patients did not necessarily present as one of the previously described distinct clinical phenotypes. DLD deficiency is a panethnic disorder, with significant phenotypic variability, and comprises a continuum, rather than three distinct clinical presentations.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
