{"title":"H-ZSM-5 沸石催化甲醇制烯烃的机理研究:DFT 研究。","authors":"Ke Pang, Ruipeng Ren, Yongkang Lv, Gui-Chang Wang","doi":"10.1007/s00894-024-06080-2","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>The mechanisms for the formation of the first C − C bond and lower olefins on methanol to olefins (MTO) conversion on H-ZSM-5 had been focused in dispute. In this paper, density functional theory has been used to study the reaction mechanisms of methanol to olefins on ZSM-5. The configurations of reactants, intermediates, products and transition state of the numerous reactions involved in such a process have been optimized, as well as the elementary reactions related to these configurations were determined by the calculation of corresponding activation energy barriers and reaction heats. Here, two different kinds of the mechanisms were proposed for the formation of dimethyl ether (DME), one involving an associative interaction of two methanol molecules with the zeolite Brønsted acid sites and the other occurring via a surface methoxy species and a methanol molecule. A critical intermediate of the methoxy methyl cation was theoretically verified by the reaction of the methoxy species and dimethyl ether. Besides, it was found that the first intermediates containing a C − C bond were 1,2-dimethoxyethane and 2-methoxy-ethanolare, in which the former was formed from methoxy species with dimethyl ether and the latter was formed from methanol by onium ions((CH<sub>3</sub>)<sub>2</sub>O<sup>+</sup>CH<sub>2</sub>CH<sub>2</sub>OCH<sub>3</sub>), respectively. For the whole reaction mechanism, the results in this paper indicated that the ethene formation is more favorable than propylene formation due to the low activation energy barrier for ethene formation (123.49 vs. 162.09 kJ.mol<sup>−1</sup>). From these calculations, it would be concluded that ethene is the first alkene product that induces the occurrence of the hydrocarbon pool mechanism.</p><h3>Methods</h3><p>All the periodic density function theory (DFT) calculations were performed by the Vienna Ab Initio Simulation package (VASP). The interaction between nucleus and valence electron was described using the pseudopotentials found in the projector augmented wave (PAW) method. PBE-D3 was used in the whole DFT calculations and CI-NEB was used to locate transition state.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 8","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2024-07-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Mechanistic investigation of methanol-to-olefins conversion catalyzed by H-ZSM-5 zeolite: a DFT study\",\"authors\":\"Ke Pang, Ruipeng Ren, Yongkang Lv, Gui-Chang Wang\",\"doi\":\"10.1007/s00894-024-06080-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>The mechanisms for the formation of the first C − C bond and lower olefins on methanol to olefins (MTO) conversion on H-ZSM-5 had been focused in dispute. In this paper, density functional theory has been used to study the reaction mechanisms of methanol to olefins on ZSM-5. The configurations of reactants, intermediates, products and transition state of the numerous reactions involved in such a process have been optimized, as well as the elementary reactions related to these configurations were determined by the calculation of corresponding activation energy barriers and reaction heats. Here, two different kinds of the mechanisms were proposed for the formation of dimethyl ether (DME), one involving an associative interaction of two methanol molecules with the zeolite Brønsted acid sites and the other occurring via a surface methoxy species and a methanol molecule. A critical intermediate of the methoxy methyl cation was theoretically verified by the reaction of the methoxy species and dimethyl ether. Besides, it was found that the first intermediates containing a C − C bond were 1,2-dimethoxyethane and 2-methoxy-ethanolare, in which the former was formed from methoxy species with dimethyl ether and the latter was formed from methanol by onium ions((CH<sub>3</sub>)<sub>2</sub>O<sup>+</sup>CH<sub>2</sub>CH<sub>2</sub>OCH<sub>3</sub>), respectively. For the whole reaction mechanism, the results in this paper indicated that the ethene formation is more favorable than propylene formation due to the low activation energy barrier for ethene formation (123.49 vs. 162.09 kJ.mol<sup>−1</sup>). From these calculations, it would be concluded that ethene is the first alkene product that induces the occurrence of the hydrocarbon pool mechanism.</p><h3>Methods</h3><p>All the periodic density function theory (DFT) calculations were performed by the Vienna Ab Initio Simulation package (VASP). The interaction between nucleus and valence electron was described using the pseudopotentials found in the projector augmented wave (PAW) method. PBE-D3 was used in the whole DFT calculations and CI-NEB was used to locate transition state.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"30 8\",\"pages\":\"\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-07-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-024-06080-2\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06080-2","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
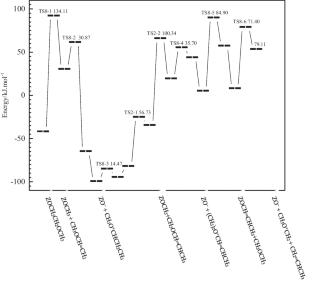
背景:甲醇在 H-ZSM-5 上转化为烯烃(MTO)时形成第一个 C - C 键和低级烯烃的机理一直是争议的焦点。本文采用密度泛函理论研究了甲醇在 ZSM-5 上转化为烯烃的反应机理。通过计算相应的活化能垒和反应热,对这一过程中涉及的众多反应的反应物、中间产物、产物和过渡态的构型进行了优化,并确定了与这些构型相关的基本反应。在这里,提出了形成二甲醚(DME)的两种不同机理,一种涉及两个甲醇分子与沸石布氏酸位点的关联作用,另一种则通过表面甲氧基物种和甲醇分子发生。甲氧基甲基阳离子的临界中间体通过甲氧基物种和二甲醚的反应得到了理论验证。此外,还发现含有 C - C 键的第一中间体是 1,2 二甲基乙烷和 2-甲氧基乙醇,其中前者由甲氧基与二甲醚生成,后者由甲醇与鎓离子((CH3)2O+CH2CH2OCH3)生成。就整个反应机理而言,本文的研究结果表明,由于乙烯形成的活化能势垒较低(123.49 对 162.09 kJ.mol-1),乙烯的形成比丙烯的形成更有利。从这些计算结果中可以得出结论,乙烯是诱导烃池机制发生的第一种烯烃产物:所有周期性密度函数理论(DFT)计算均由维也纳 Ab Initio 仿真软件包(VASP)完成。原子核与价电子之间的相互作用是用投影增强波(PAW)方法中的伪势来描述的。在整个 DFT 计算中使用了 PBE-D3,并使用 CI-NEB 定位过渡态。
Mechanistic investigation of methanol-to-olefins conversion catalyzed by H-ZSM-5 zeolite: a DFT study
Context
The mechanisms for the formation of the first C − C bond and lower olefins on methanol to olefins (MTO) conversion on H-ZSM-5 had been focused in dispute. In this paper, density functional theory has been used to study the reaction mechanisms of methanol to olefins on ZSM-5. The configurations of reactants, intermediates, products and transition state of the numerous reactions involved in such a process have been optimized, as well as the elementary reactions related to these configurations were determined by the calculation of corresponding activation energy barriers and reaction heats. Here, two different kinds of the mechanisms were proposed for the formation of dimethyl ether (DME), one involving an associative interaction of two methanol molecules with the zeolite Brønsted acid sites and the other occurring via a surface methoxy species and a methanol molecule. A critical intermediate of the methoxy methyl cation was theoretically verified by the reaction of the methoxy species and dimethyl ether. Besides, it was found that the first intermediates containing a C − C bond were 1,2-dimethoxyethane and 2-methoxy-ethanolare, in which the former was formed from methoxy species with dimethyl ether and the latter was formed from methanol by onium ions((CH3)2O+CH2CH2OCH3), respectively. For the whole reaction mechanism, the results in this paper indicated that the ethene formation is more favorable than propylene formation due to the low activation energy barrier for ethene formation (123.49 vs. 162.09 kJ.mol−1). From these calculations, it would be concluded that ethene is the first alkene product that induces the occurrence of the hydrocarbon pool mechanism.
Methods
All the periodic density function theory (DFT) calculations were performed by the Vienna Ab Initio Simulation package (VASP). The interaction between nucleus and valence electron was described using the pseudopotentials found in the projector augmented wave (PAW) method. PBE-D3 was used in the whole DFT calculations and CI-NEB was used to locate transition state.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
