{"title":"通过计算方法设计和发现单极纺锤体激酶 1 (MPS1/TTK) 抑制剂","authors":"Nan Li, Jianning Wang, Haiyue Wu, Zhichao Zheng, Wei Liu, Zijian Qin","doi":"10.1007/s00044-024-03286-0","DOIUrl":null,"url":null,"abstract":"<div><p>Monopolar spindle kinase 1 (MPS1, also called TTK) is an attractive target for the treatment of cancers. Five MPS1 inhibitors have entered the clinical trials, but four of them were discontinued; thus, it is necessary to develop MPS1 inhibitors with novel scaffolds. In the present work, several computational tools were built to design MPS1 inhibitors. The deep recurrent neural network was used for generating potential highly active MPS1 inhibitors. The deep neural network was used to build a ligand-based consensus model for distinguishing the highly and weakly active MPS1 inhibitors. Five co-crystal structures were used to develop the consensus docking score for distinguishing actives and decoys. The ligand-based consensus model and the consensus docking score were used to evaluate the generated molecules, and finally, two scaffolds, which were less reported as MPS1 inhibitors previously, were selected. A total of 15 compounds with the two scaffolds were synthesized and tested by in vitro enzymatic inhibition assays. Five compounds had sub-micromolar to low micromolar in vitro potencies, and the most active compound was <b>10</b> with an IC<sub>50</sub> of 556 nM. The binding modes of the new compounds were revealed by molecular dynamic simulations. We believe that the computational strategies in the present work were helpful for discovering new potential scaffolds.</p><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":699,"journal":{"name":"Medicinal Chemistry Research","volume":"33 10","pages":"1861 - 1886"},"PeriodicalIF":3.1000,"publicationDate":"2024-07-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Design and discovery of monopolar spindle kinase 1 (MPS1/TTK) inhibitors by computational approaches\",\"authors\":\"Nan Li, Jianning Wang, Haiyue Wu, Zhichao Zheng, Wei Liu, Zijian Qin\",\"doi\":\"10.1007/s00044-024-03286-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Monopolar spindle kinase 1 (MPS1, also called TTK) is an attractive target for the treatment of cancers. Five MPS1 inhibitors have entered the clinical trials, but four of them were discontinued; thus, it is necessary to develop MPS1 inhibitors with novel scaffolds. In the present work, several computational tools were built to design MPS1 inhibitors. The deep recurrent neural network was used for generating potential highly active MPS1 inhibitors. The deep neural network was used to build a ligand-based consensus model for distinguishing the highly and weakly active MPS1 inhibitors. Five co-crystal structures were used to develop the consensus docking score for distinguishing actives and decoys. The ligand-based consensus model and the consensus docking score were used to evaluate the generated molecules, and finally, two scaffolds, which were less reported as MPS1 inhibitors previously, were selected. A total of 15 compounds with the two scaffolds were synthesized and tested by in vitro enzymatic inhibition assays. Five compounds had sub-micromolar to low micromolar in vitro potencies, and the most active compound was <b>10</b> with an IC<sub>50</sub> of 556 nM. The binding modes of the new compounds were revealed by molecular dynamic simulations. We believe that the computational strategies in the present work were helpful for discovering new potential scaffolds.</p><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>\",\"PeriodicalId\":699,\"journal\":{\"name\":\"Medicinal Chemistry Research\",\"volume\":\"33 10\",\"pages\":\"1861 - 1886\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-07-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Medicinal Chemistry Research\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00044-024-03286-0\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Medicinal Chemistry Research","FirstCategoryId":"3","ListUrlMain":"https://link.springer.com/article/10.1007/s00044-024-03286-0","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Design and discovery of monopolar spindle kinase 1 (MPS1/TTK) inhibitors by computational approaches
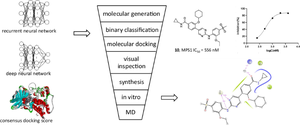
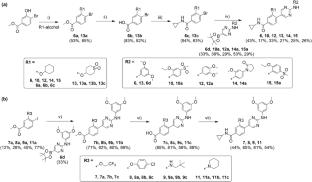
Monopolar spindle kinase 1 (MPS1, also called TTK) is an attractive target for the treatment of cancers. Five MPS1 inhibitors have entered the clinical trials, but four of them were discontinued; thus, it is necessary to develop MPS1 inhibitors with novel scaffolds. In the present work, several computational tools were built to design MPS1 inhibitors. The deep recurrent neural network was used for generating potential highly active MPS1 inhibitors. The deep neural network was used to build a ligand-based consensus model for distinguishing the highly and weakly active MPS1 inhibitors. Five co-crystal structures were used to develop the consensus docking score for distinguishing actives and decoys. The ligand-based consensus model and the consensus docking score were used to evaluate the generated molecules, and finally, two scaffolds, which were less reported as MPS1 inhibitors previously, were selected. A total of 15 compounds with the two scaffolds were synthesized and tested by in vitro enzymatic inhibition assays. Five compounds had sub-micromolar to low micromolar in vitro potencies, and the most active compound was 10 with an IC50 of 556 nM. The binding modes of the new compounds were revealed by molecular dynamic simulations. We believe that the computational strategies in the present work were helpful for discovering new potential scaffolds.
期刊介绍:
Medicinal Chemistry Research (MCRE) publishes papers on a wide range of topics, favoring research with significant, new, and up-to-date information. Although the journal has a demanding peer review process, MCRE still boasts rapid publication, due in part, to the length of the submissions. The journal publishes significant research on various topics, many of which emphasize the structure-activity relationships of molecular biology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
