{"title":"利用蛋白质质谱进行结构系统学研究:概念验证。","authors":"Benjamin P. Todd, Kevin M. Downard","doi":"10.1007/s10930-024-10227-8","DOIUrl":null,"url":null,"abstract":"<div><p>It is demonstrated, for the first time, that a mass spectrometry approach (known as <i>phylonumerics</i>) can be successfully implemented for structural phylogenetics investigations to chart the evolution of a protein’s structure and function. Illustrated for the compact globular protein myoglobin, peptide masses produced from the proteolytic digestion of the protein across animal species generate trees congruent to the sequence tree counterparts. Single point mutations calculated during the same mass tree building step can be followed along interconnected branches of the tree and represent a viable structural metric. A mass tree built for 15 diverse animal species, easily resolve the birds from mammal species, and the ruminant mammals from the remainder of the animals. Mutations within helix-spanning peptide segments alter both the mass and structure of the protein in these segments. Greater evolution is found in the B-helix over the A, E, F, G and H helices. A further mass tree study, of six more closely related primate species, resolves gorilla from the other primates based on a P22S mutation within the B-helix. The remaining five primates are resolved into two groups based on whether they contain a glycine or serine at position 23 in the same helix. The orangutan is resolved from the gibbon and siamang by its G-helix C110S mutation, while homo sapiens are resolved from chimpanzee based on the Q116H mutation. All are associated with structural perturbations in such helices. These structure altering mutations can be tracked along interconnecting branches of a mass tree, to follow the protein’s structure and evolution, and ultimately the evolution of the species in which the proteins are expressed. Those that have the greatest impact on a protein’s structure, its function, and ultimately the evolution of the species, can be selectively tracked or monitored.</p></div>","PeriodicalId":793,"journal":{"name":"The Protein Journal","volume":"43 5","pages":"997 - 1008"},"PeriodicalIF":1.4000,"publicationDate":"2024-07-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Structural Phylogenetics with Protein Mass Spectrometry: A Proof-of-Concept\",\"authors\":\"Benjamin P. Todd, Kevin M. Downard\",\"doi\":\"10.1007/s10930-024-10227-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>It is demonstrated, for the first time, that a mass spectrometry approach (known as <i>phylonumerics</i>) can be successfully implemented for structural phylogenetics investigations to chart the evolution of a protein’s structure and function. Illustrated for the compact globular protein myoglobin, peptide masses produced from the proteolytic digestion of the protein across animal species generate trees congruent to the sequence tree counterparts. Single point mutations calculated during the same mass tree building step can be followed along interconnected branches of the tree and represent a viable structural metric. A mass tree built for 15 diverse animal species, easily resolve the birds from mammal species, and the ruminant mammals from the remainder of the animals. Mutations within helix-spanning peptide segments alter both the mass and structure of the protein in these segments. Greater evolution is found in the B-helix over the A, E, F, G and H helices. A further mass tree study, of six more closely related primate species, resolves gorilla from the other primates based on a P22S mutation within the B-helix. The remaining five primates are resolved into two groups based on whether they contain a glycine or serine at position 23 in the same helix. The orangutan is resolved from the gibbon and siamang by its G-helix C110S mutation, while homo sapiens are resolved from chimpanzee based on the Q116H mutation. All are associated with structural perturbations in such helices. These structure altering mutations can be tracked along interconnecting branches of a mass tree, to follow the protein’s structure and evolution, and ultimately the evolution of the species in which the proteins are expressed. Those that have the greatest impact on a protein’s structure, its function, and ultimately the evolution of the species, can be selectively tracked or monitored.</p></div>\",\"PeriodicalId\":793,\"journal\":{\"name\":\"The Protein Journal\",\"volume\":\"43 5\",\"pages\":\"997 - 1008\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2024-07-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Protein Journal\",\"FirstCategoryId\":\"2\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10930-024-10227-8\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Protein Journal","FirstCategoryId":"2","ListUrlMain":"https://link.springer.com/article/10.1007/s10930-024-10227-8","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
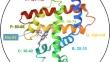
该研究首次证明,质谱方法(称为 "系统数值学")可成功应用于结构系统学研究,以绘制蛋白质结构和功能的进化图谱。以结构紧凑的球状蛋白质肌红蛋白为例,通过对不同动物物种的蛋白质进行蛋白酶解产生的肽质量生成了与序列树对应的树。在同一质量树构建步骤中计算出的单点突变可沿着质量树相互连接的分支进行追踪,是一种可行的结构度量方法。为 15 个不同动物物种构建的质量树可以很容易地将鸟类与哺乳动物物种区分开来,并将反刍哺乳动物与其他动物区分开来。跨越螺旋的肽段内的突变会改变这些肽段内蛋白质的质量和结构。与 A、E、F、G 和 H 螺旋相比,B 螺旋的进化程度更高。对六种亲缘关系更近的灵长类动物进行的进一步质量树研究,根据 B 螺旋中的 P22S 突变,将大猩猩与其他灵长类动物区分开来。其余五种灵长类动物则根据它们在同一螺旋的第 23 位含有甘氨酸还是丝氨酸分成了两组。猩猩因其 G 螺旋 C110S 突变而与长臂猿和暹罗猿区分开来,而智人则因 Q116H 突变而与黑猩猩区分开来。所有这些都与这些螺旋的结构扰动有关。这些改变结构的突变可以沿着质量树相互连接的分支进行追踪,以了解蛋白质的结构和进化,并最终了解表达蛋白质的物种的进化。可以有选择地跟踪或监测那些对蛋白质结构、功能以及物种进化影响最大的突变。
Structural Phylogenetics with Protein Mass Spectrometry: A Proof-of-Concept
It is demonstrated, for the first time, that a mass spectrometry approach (known as phylonumerics) can be successfully implemented for structural phylogenetics investigations to chart the evolution of a protein’s structure and function. Illustrated for the compact globular protein myoglobin, peptide masses produced from the proteolytic digestion of the protein across animal species generate trees congruent to the sequence tree counterparts. Single point mutations calculated during the same mass tree building step can be followed along interconnected branches of the tree and represent a viable structural metric. A mass tree built for 15 diverse animal species, easily resolve the birds from mammal species, and the ruminant mammals from the remainder of the animals. Mutations within helix-spanning peptide segments alter both the mass and structure of the protein in these segments. Greater evolution is found in the B-helix over the A, E, F, G and H helices. A further mass tree study, of six more closely related primate species, resolves gorilla from the other primates based on a P22S mutation within the B-helix. The remaining five primates are resolved into two groups based on whether they contain a glycine or serine at position 23 in the same helix. The orangutan is resolved from the gibbon and siamang by its G-helix C110S mutation, while homo sapiens are resolved from chimpanzee based on the Q116H mutation. All are associated with structural perturbations in such helices. These structure altering mutations can be tracked along interconnecting branches of a mass tree, to follow the protein’s structure and evolution, and ultimately the evolution of the species in which the proteins are expressed. Those that have the greatest impact on a protein’s structure, its function, and ultimately the evolution of the species, can be selectively tracked or monitored.
期刊介绍:
The Protein Journal (formerly the Journal of Protein Chemistry) publishes original research work on all aspects of proteins and peptides. These include studies concerned with covalent or three-dimensional structure determination (X-ray, NMR, cryoEM, EPR/ESR, optical methods, etc.), computational aspects of protein structure and function, protein folding and misfolding, assembly, genetics, evolution, proteomics, molecular biology, protein engineering, protein nanotechnology, protein purification and analysis and peptide synthesis, as well as the elucidation and interpretation of the molecular bases of biological activities of proteins and peptides. We accept original research papers, reviews, mini-reviews, hypotheses, opinion papers, and letters to the editor.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
