Petri Pölönen, Danika Di Giacomo, Anna Eames Seffernick, Abdelrahman Elsayed, Shunsuke Kimura, Francesca Benini, Lindsey E. Montefiori, Brent L. Wood, Jason Xu, Changya Chen, Zhongshan Cheng, Haley Newman, Jason Myers, Ilaria Iacobucci, Elizabeth Li, Jonathan Sussman, Dale Hedges, Yawei Hui, Caroline Diorio, Lahari Uppuluri, David Frank, Yiping Fan, Yunchao Chang, Soheil Meshinchi, Rhonda Ries, Rawan Shraim, Alexander Li, Kathrin M. Bernt, Meenakshi Devidas, Stuart S. Winter, Kimberly P. Dunsmore, Hiroto Inaba, William L. Carroll, Nilsa C. Ramirez, Aaron H. Phillips, Richard W. Kriwacki, Jun J. Yang, Tiffaney L. Vincent, Yaqi Zhao, Pankaj S. Ghate, Jian Wang, Colleen Reilly, Xin Zhou, Mathijs A. Sanders, Junko Takita, Motohiro Kato, Nao Takasugi, Bill H. Chang, Richard D. Press, Mignon Loh, Evadnie Rampersaud, Elizabeth Raetz, Stephen P. Hunger, Kai Tan, Ti-Cheng Chang, Gang Wu, Stanley B. Pounds, Charles G. Mullighan, David T. Teachey
{"title":"儿童 T 线型急性淋巴细胞白血病的基因组基础。","authors":"Petri Pölönen, Danika Di Giacomo, Anna Eames Seffernick, Abdelrahman Elsayed, Shunsuke Kimura, Francesca Benini, Lindsey E. Montefiori, Brent L. Wood, Jason Xu, Changya Chen, Zhongshan Cheng, Haley Newman, Jason Myers, Ilaria Iacobucci, Elizabeth Li, Jonathan Sussman, Dale Hedges, Yawei Hui, Caroline Diorio, Lahari Uppuluri, David Frank, Yiping Fan, Yunchao Chang, Soheil Meshinchi, Rhonda Ries, Rawan Shraim, Alexander Li, Kathrin M. Bernt, Meenakshi Devidas, Stuart S. Winter, Kimberly P. Dunsmore, Hiroto Inaba, William L. Carroll, Nilsa C. Ramirez, Aaron H. Phillips, Richard W. Kriwacki, Jun J. Yang, Tiffaney L. Vincent, Yaqi Zhao, Pankaj S. Ghate, Jian Wang, Colleen Reilly, Xin Zhou, Mathijs A. Sanders, Junko Takita, Motohiro Kato, Nao Takasugi, Bill H. Chang, Richard D. Press, Mignon Loh, Evadnie Rampersaud, Elizabeth Raetz, Stephen P. Hunger, Kai Tan, Ti-Cheng Chang, Gang Wu, Stanley B. Pounds, Charles G. Mullighan, David T. Teachey","doi":"10.1038/s41586-024-07807-0","DOIUrl":null,"url":null,"abstract":"T-lineage acute lymphoblastic leukaemia (T-ALL) is a high-risk tumour1 that has eluded comprehensive genomic characterization, which is partly due to the high frequency of noncoding genomic alterations that result in oncogene deregulation2,3. Here we report an integrated analysis of genome and transcriptome sequencing of tumour and remission samples from more than 1,300 uniformly treated children with T-ALL, coupled with epigenomic and single-cell analyses of malignant and normal T cell precursors. This approach identified 15 subtypes with distinct genomic drivers, gene expression patterns, developmental states and outcomes. Analyses of chromatin topology revealed multiple mechanisms of enhancer deregulation that involve enhancers and genes in a subtype-specific manner, thereby demonstrating widespread involvement of the noncoding genome. We show that the immunophenotypically described, high-risk entity of early T cell precursor ALL is superseded by a broader category of ‘early T cell precursor-like’ leukaemia. This category has a variable immunophenotype and diverse genomic alterations of a core set of genes that encode regulators of hematopoietic stem cell development. Using multivariable outcome models, we show that genetic subtypes, driver and concomitant genetic alterations independently predict treatment failure and survival. These findings provide a roadmap for the classification, risk stratification and mechanistic understanding of this disease. Comprehensive genomic and transcriptomics analyses of more than 1,300 cases of childhood T-lineage acute lymphoblastic leukaemia identify 15 distinct subtypes that are associated with specific outcomes.","PeriodicalId":18787,"journal":{"name":"Nature","volume":"632 8027","pages":"1082-1091"},"PeriodicalIF":48.5000,"publicationDate":"2024-08-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The genomic basis of childhood T-lineage acute lymphoblastic leukaemia\",\"authors\":\"Petri Pölönen, Danika Di Giacomo, Anna Eames Seffernick, Abdelrahman Elsayed, Shunsuke Kimura, Francesca Benini, Lindsey E. Montefiori, Brent L. Wood, Jason Xu, Changya Chen, Zhongshan Cheng, Haley Newman, Jason Myers, Ilaria Iacobucci, Elizabeth Li, Jonathan Sussman, Dale Hedges, Yawei Hui, Caroline Diorio, Lahari Uppuluri, David Frank, Yiping Fan, Yunchao Chang, Soheil Meshinchi, Rhonda Ries, Rawan Shraim, Alexander Li, Kathrin M. Bernt, Meenakshi Devidas, Stuart S. Winter, Kimberly P. Dunsmore, Hiroto Inaba, William L. Carroll, Nilsa C. Ramirez, Aaron H. Phillips, Richard W. Kriwacki, Jun J. Yang, Tiffaney L. Vincent, Yaqi Zhao, Pankaj S. Ghate, Jian Wang, Colleen Reilly, Xin Zhou, Mathijs A. Sanders, Junko Takita, Motohiro Kato, Nao Takasugi, Bill H. Chang, Richard D. Press, Mignon Loh, Evadnie Rampersaud, Elizabeth Raetz, Stephen P. Hunger, Kai Tan, Ti-Cheng Chang, Gang Wu, Stanley B. Pounds, Charles G. Mullighan, David T. Teachey\",\"doi\":\"10.1038/s41586-024-07807-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"T-lineage acute lymphoblastic leukaemia (T-ALL) is a high-risk tumour1 that has eluded comprehensive genomic characterization, which is partly due to the high frequency of noncoding genomic alterations that result in oncogene deregulation2,3. Here we report an integrated analysis of genome and transcriptome sequencing of tumour and remission samples from more than 1,300 uniformly treated children with T-ALL, coupled with epigenomic and single-cell analyses of malignant and normal T cell precursors. This approach identified 15 subtypes with distinct genomic drivers, gene expression patterns, developmental states and outcomes. Analyses of chromatin topology revealed multiple mechanisms of enhancer deregulation that involve enhancers and genes in a subtype-specific manner, thereby demonstrating widespread involvement of the noncoding genome. We show that the immunophenotypically described, high-risk entity of early T cell precursor ALL is superseded by a broader category of ‘early T cell precursor-like’ leukaemia. This category has a variable immunophenotype and diverse genomic alterations of a core set of genes that encode regulators of hematopoietic stem cell development. Using multivariable outcome models, we show that genetic subtypes, driver and concomitant genetic alterations independently predict treatment failure and survival. These findings provide a roadmap for the classification, risk stratification and mechanistic understanding of this disease. Comprehensive genomic and transcriptomics analyses of more than 1,300 cases of childhood T-lineage acute lymphoblastic leukaemia identify 15 distinct subtypes that are associated with specific outcomes.\",\"PeriodicalId\":18787,\"journal\":{\"name\":\"Nature\",\"volume\":\"632 8027\",\"pages\":\"1082-1091\"},\"PeriodicalIF\":48.5000,\"publicationDate\":\"2024-08-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature\",\"FirstCategoryId\":\"103\",\"ListUrlMain\":\"https://www.nature.com/articles/s41586-024-07807-0\",\"RegionNum\":1,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature","FirstCategoryId":"103","ListUrlMain":"https://www.nature.com/articles/s41586-024-07807-0","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
摘要
T系急性淋巴细胞白血病(T-ALL)是一种高危肿瘤1,一直未能得到全面的基因组学鉴定,部分原因是非编码基因组的高频率改变导致了癌基因的失调2,3。在此,我们报告了对 1300 多名接受过统一治疗的 T-ALL 儿童的肿瘤和缓解样本进行基因组和转录组测序的综合分析,以及对恶性和正常 T 细胞前体进行表观基因组和单细胞分析的结果。这种方法确定了具有不同基因组驱动因素、基因表达模式、发育状态和结果的15种亚型。染色质拓扑分析揭示了增强子失调的多种机制,这些机制以亚型特异的方式涉及增强子和基因,从而证明了非编码基因组的广泛参与。我们发现,免疫表型上描述的早期 T 细胞前体 ALL 这一高风险实体已被更广泛的 "早期 T 细胞前体样 "白血病类别所取代。这类白血病的免疫表型多变,一组编码造血干细胞发育调控因子的核心基因发生了不同的基因组改变。利用多变量结果模型,我们发现基因亚型、驱动基因和伴随基因改变可独立预测治疗失败和生存期。这些发现为这种疾病的分类、风险分层和机理理解提供了路线图。
The genomic basis of childhood T-lineage acute lymphoblastic leukaemia
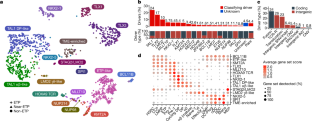
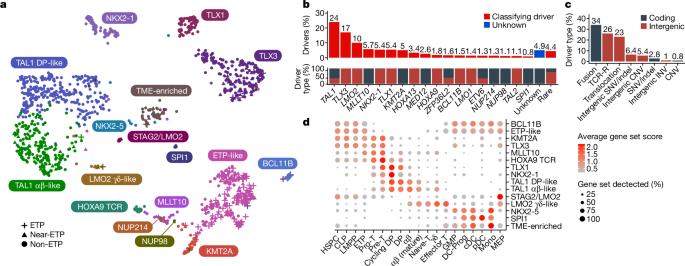
T-lineage acute lymphoblastic leukaemia (T-ALL) is a high-risk tumour1 that has eluded comprehensive genomic characterization, which is partly due to the high frequency of noncoding genomic alterations that result in oncogene deregulation2,3. Here we report an integrated analysis of genome and transcriptome sequencing of tumour and remission samples from more than 1,300 uniformly treated children with T-ALL, coupled with epigenomic and single-cell analyses of malignant and normal T cell precursors. This approach identified 15 subtypes with distinct genomic drivers, gene expression patterns, developmental states and outcomes. Analyses of chromatin topology revealed multiple mechanisms of enhancer deregulation that involve enhancers and genes in a subtype-specific manner, thereby demonstrating widespread involvement of the noncoding genome. We show that the immunophenotypically described, high-risk entity of early T cell precursor ALL is superseded by a broader category of ‘early T cell precursor-like’ leukaemia. This category has a variable immunophenotype and diverse genomic alterations of a core set of genes that encode regulators of hematopoietic stem cell development. Using multivariable outcome models, we show that genetic subtypes, driver and concomitant genetic alterations independently predict treatment failure and survival. These findings provide a roadmap for the classification, risk stratification and mechanistic understanding of this disease. Comprehensive genomic and transcriptomics analyses of more than 1,300 cases of childhood T-lineage acute lymphoblastic leukaemia identify 15 distinct subtypes that are associated with specific outcomes.
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
