{"title":"PMiSLocMF:通过结合 miRNA 的多源特征预测 miRNA 的亚细胞定位。","authors":"Lei Chen, Jiahui Gu, Bo Zhou","doi":"10.1093/bib/bbae386","DOIUrl":null,"url":null,"abstract":"<p><p>The microRNAs (miRNAs) play crucial roles in several biological processes. It is essential for a deeper insight into their functions and mechanisms by detecting their subcellular localizations. The traditional methods for determining miRNAs subcellular localizations are expensive. The computational methods are alternative ways to quickly predict miRNAs subcellular localizations. Although several computational methods have been proposed in this regard, the incomplete representations of miRNAs in these methods left the room for improvement. In this study, a novel computational method for predicting miRNA subcellular localizations, named PMiSLocMF, was developed. As lots of miRNAs have multiple subcellular localizations, this method was a multi-label classifier. Several properties of miRNA, such as miRNA sequences, miRNA functional similarity, miRNA-disease, miRNA-drug, and miRNA-mRNA associations were adopted for generating informative miRNA features. To this end, powerful algorithms [node2vec and graph attention auto-encoder (GATE)] and one newly designed scheme were adopted to process above properties, producing five feature types. All features were poured into self-attention and fully connected layers to make predictions. The cross-validation results indicated the high performance of PMiSLocMF with accuracy higher than 0.83, average area under the receiver operating characteristic curve (AUC) and area under the precision-recall curve (AUPR) exceeding 0.90 and 0.77, respectively. Such performance was better than all previous methods based on the same dataset. Further tests proved that using all feature types can improve the performance of PMiSLocMF, and GATE and self-attention layer can help enhance the performance. Finally, we deeply analyzed the influence of miRNA associations with diseases, drugs, and mRNAs on PMiSLocMF. The dataset and codes are available at https://github.com/Gu20201017/PMiSLocMF.</p>","PeriodicalId":9209,"journal":{"name":"Briefings in bioinformatics","volume":"25 5","pages":""},"PeriodicalIF":7.7000,"publicationDate":"2024-07-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11330342/pdf/","citationCount":"0","resultStr":"{\"title\":\"PMiSLocMF: predicting miRNA subcellular localizations by incorporating multi-source features of miRNAs.\",\"authors\":\"Lei Chen, Jiahui Gu, Bo Zhou\",\"doi\":\"10.1093/bib/bbae386\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The microRNAs (miRNAs) play crucial roles in several biological processes. It is essential for a deeper insight into their functions and mechanisms by detecting their subcellular localizations. The traditional methods for determining miRNAs subcellular localizations are expensive. The computational methods are alternative ways to quickly predict miRNAs subcellular localizations. Although several computational methods have been proposed in this regard, the incomplete representations of miRNAs in these methods left the room for improvement. In this study, a novel computational method for predicting miRNA subcellular localizations, named PMiSLocMF, was developed. As lots of miRNAs have multiple subcellular localizations, this method was a multi-label classifier. Several properties of miRNA, such as miRNA sequences, miRNA functional similarity, miRNA-disease, miRNA-drug, and miRNA-mRNA associations were adopted for generating informative miRNA features. To this end, powerful algorithms [node2vec and graph attention auto-encoder (GATE)] and one newly designed scheme were adopted to process above properties, producing five feature types. All features were poured into self-attention and fully connected layers to make predictions. The cross-validation results indicated the high performance of PMiSLocMF with accuracy higher than 0.83, average area under the receiver operating characteristic curve (AUC) and area under the precision-recall curve (AUPR) exceeding 0.90 and 0.77, respectively. Such performance was better than all previous methods based on the same dataset. Further tests proved that using all feature types can improve the performance of PMiSLocMF, and GATE and self-attention layer can help enhance the performance. Finally, we deeply analyzed the influence of miRNA associations with diseases, drugs, and mRNAs on PMiSLocMF. The dataset and codes are available at https://github.com/Gu20201017/PMiSLocMF.</p>\",\"PeriodicalId\":9209,\"journal\":{\"name\":\"Briefings in bioinformatics\",\"volume\":\"25 5\",\"pages\":\"\"},\"PeriodicalIF\":7.7000,\"publicationDate\":\"2024-07-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11330342/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Briefings in bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bib/bbae386\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bib/bbae386","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
PMiSLocMF: predicting miRNA subcellular localizations by incorporating multi-source features of miRNAs.
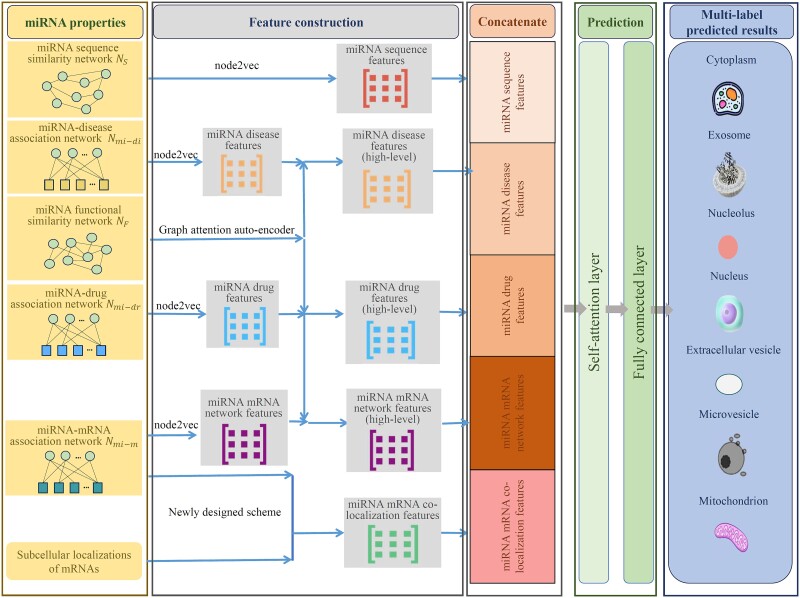
The microRNAs (miRNAs) play crucial roles in several biological processes. It is essential for a deeper insight into their functions and mechanisms by detecting their subcellular localizations. The traditional methods for determining miRNAs subcellular localizations are expensive. The computational methods are alternative ways to quickly predict miRNAs subcellular localizations. Although several computational methods have been proposed in this regard, the incomplete representations of miRNAs in these methods left the room for improvement. In this study, a novel computational method for predicting miRNA subcellular localizations, named PMiSLocMF, was developed. As lots of miRNAs have multiple subcellular localizations, this method was a multi-label classifier. Several properties of miRNA, such as miRNA sequences, miRNA functional similarity, miRNA-disease, miRNA-drug, and miRNA-mRNA associations were adopted for generating informative miRNA features. To this end, powerful algorithms [node2vec and graph attention auto-encoder (GATE)] and one newly designed scheme were adopted to process above properties, producing five feature types. All features were poured into self-attention and fully connected layers to make predictions. The cross-validation results indicated the high performance of PMiSLocMF with accuracy higher than 0.83, average area under the receiver operating characteristic curve (AUC) and area under the precision-recall curve (AUPR) exceeding 0.90 and 0.77, respectively. Such performance was better than all previous methods based on the same dataset. Further tests proved that using all feature types can improve the performance of PMiSLocMF, and GATE and self-attention layer can help enhance the performance. Finally, we deeply analyzed the influence of miRNA associations with diseases, drugs, and mRNAs on PMiSLocMF. The dataset and codes are available at https://github.com/Gu20201017/PMiSLocMF.
期刊介绍:
Briefings in Bioinformatics is an international journal serving as a platform for researchers and educators in the life sciences. It also appeals to mathematicians, statisticians, and computer scientists applying their expertise to biological challenges. The journal focuses on reviews tailored for users of databases and analytical tools in contemporary genetics, molecular and systems biology. It stands out by offering practical assistance and guidance to non-specialists in computerized methodologies. Covering a wide range from introductory concepts to specific protocols and analyses, the papers address bacterial, plant, fungal, animal, and human data.
The journal's detailed subject areas include genetic studies of phenotypes and genotypes, mapping, DNA sequencing, expression profiling, gene expression studies, microarrays, alignment methods, protein profiles and HMMs, lipids, metabolic and signaling pathways, structure determination and function prediction, phylogenetic studies, and education and training.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
